

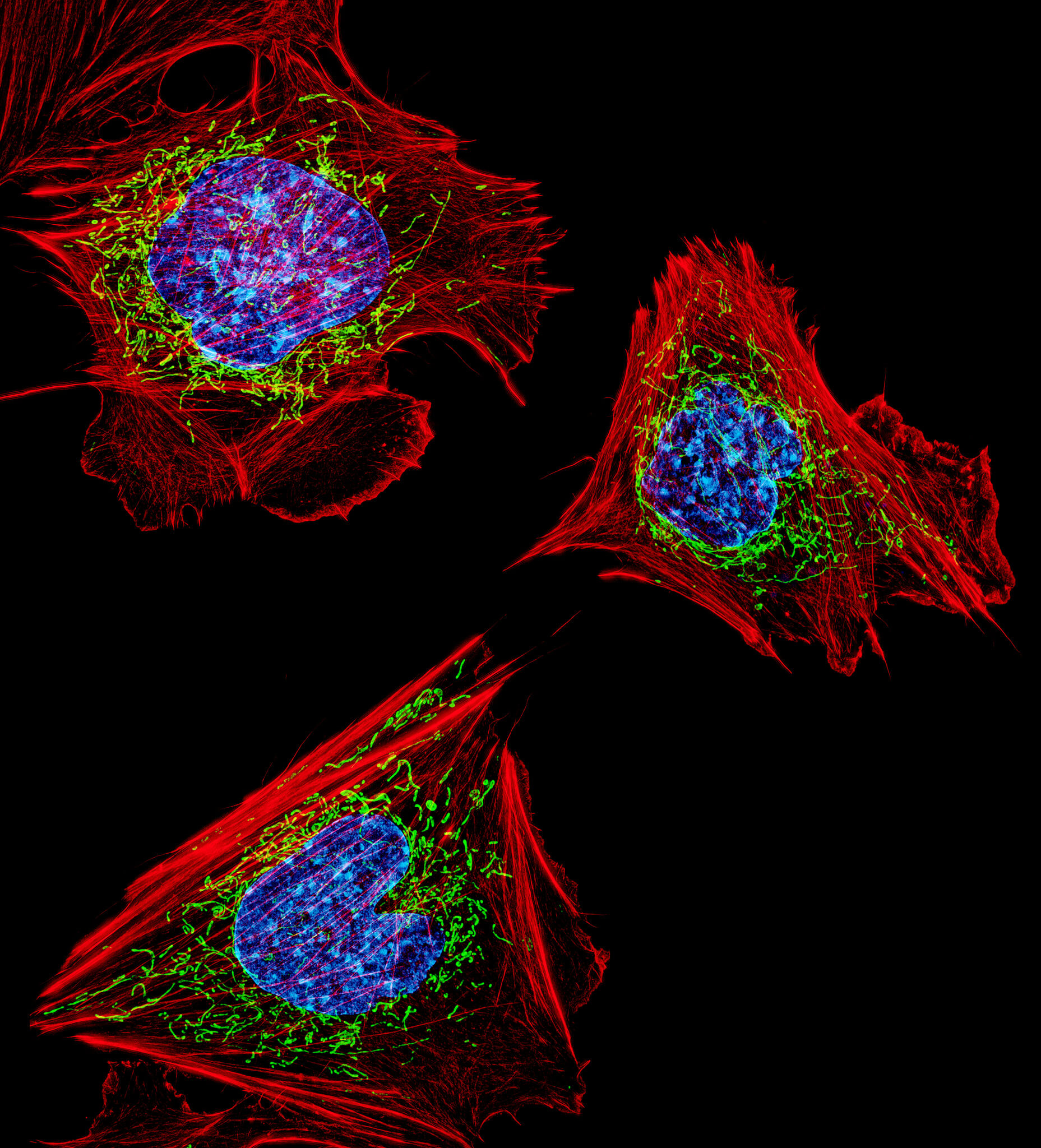
How to stress a cell
As we saw in the previous section, there are several types of stress and the cell has various responses to deal with them. The most typical responses involve distinct organelles including mitochondria, which play a central role in regulating apoptosis; the reticulum, which allows the mediation of UPR (Unfolded Protein Response); the nucleus, in which DNA lesions can trigger stress, or peroxisomes, which have an important role in the response to oxidative stress.
Let’s take a look at all these mechanisms to better understand how cellular stress can impact our cells!
Mitochondria: the first targets of cellular stress
The mitochondria are somewhat odd organelles. Their presence in the cytoplasm of our cells is the result of a fusion of an old bacterium with our cells (endosymbiotic theory) more than 2 billion years ago[1]. they have their own DNA (called mtDNA for mitochondrial DNA) and are transmitted from a mother cell to a daughter cell by increasing their size followed by fission. They have a central role in our energy metabolism since they manage the production of ATP and NADH in particular, two cofactors essential to the functioning of our enzymes. They are transmitted only by the mother, thanks to the mitochondria present in the eggs, the mitochondria of the spermatozoa not passing the membrane of the oocyte during fertilization[1]. There are mitochondrial diseases that can lead to mental delays, metabolic dysfunctions or heart problems, always of maternal origin.
In a physiological context, mitochondria play different roles and are the preferred (and most studied) target of cellular stress.
Oxidative phosphorylation and coenzymes
When a cell functions normally, it uses coenzymes (or cofactors), small molecules allowing the action of enzymes. These coenzymes include ATP and NADH. When enzymes work, they use these cofactors to bring enough energy to the reaction that they make possible and allow its acceleration. During this phenomenon, the ATP is transformed into ADP (it loses a phosphate group) and in order to recycle it, the mitochondria will implement a whole system. Without going into the details of complex metabolic processes, the mitochondria will add a phosphate group to the ADP to transform it into ATP and allow its reuse. This action is made possible by successive oxidation-reduction mechanisms that use the energy released by the use of NADH (transformed into NAD+) and the action of ATP-synthase.
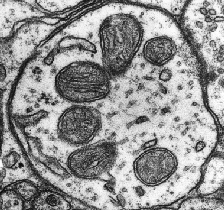
Although essential for cellular function, oxidative phosphorylation causes the formation of free radicals and reactive oxygen species (ROS)[2]. Three major forms of ROS are observed: hydrogen peroxide (H2O2), the radical -OH and the superoxide ion O2–[3]. Of these three species, the most reactive is -OH, which can modify nuclear and mitochondrial DNA and break its strands[3]. To combat this oxidative stress, our cells will develop various strategies involving antioxidant machinery (see part 1: the different types of cellular stress)[4]. Normally, the mitochondria and the cell are able to maintain a balance between the formation of these molecules and their elimination. However, during an external event or exposure to drugs known to increase ROS synthesis, this balance can tip in the wrong direction and produce oxidative cellular stress[5].
Apoptosis regulation
Apoptosis is a programmed cell death mechanism triggered by the cell when the conditions guaranteeing its survival are no longer met. It can be activated by different phenomena, but the central regulator of apoptosis remains mitochondria. The activation of the apoptotic signal following cellular stress takes place via the caspase pathway, and more particularly caspases 3, 8 and 9. Caspase-8 is the first to be activated, thanks to a complex but well-known signalling pathway involving kinases (a specific class of enzymes) including Akt, ERK1/2, p38 and JNK[6] themselves activated by proteins of the Bcl-2 family.
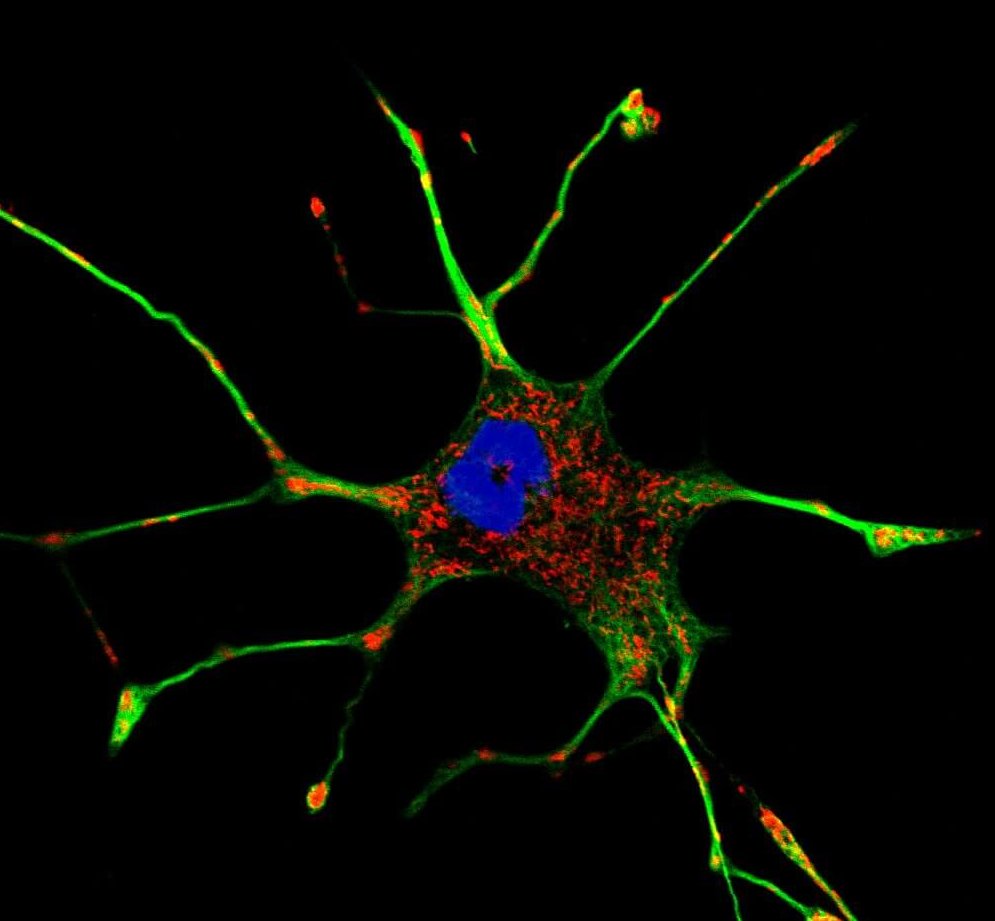
Once activated, caspase-8 will transmit the signal either directly, via caspase-3, or indirectly via the mitochondria. The mitochondria will indeed be able to react to caspase-8, which will indirectly trigger the permeabilization of its membrane[7]. The cytochrome c will be able to emerge from the mitochondria, where it is normally sequestered, and engage new pro-apoptotic pathways. The synthesis of ROS is simultaneously increased, creating a loop promoting cell death[8]. In addition to these mechanisms, molecules triggering cellular stress can directly cause permeabilization of the mitochondrial membrane, without going through caspase-8 activation. The presence of small holes (pores) in the mitochondria will disturb the membrane potential and cause a loss of cellular homeostasis: ATP synthesis will no longer take place, NADH will be oxidized and ROS will be synthesized in excess. In response to these phenomena, mitochondria will release several proteins, the best known of which is AIF (Apoptosis-Inducing Factor), which will bind at the DNA level to cleave and cause apoptosis[9].
The number of stimuli that can trigger a mitochondrial response to cellular stress is impressive. Years of research have of course helped to explain some of these mechanisms, and we now know the predominant role of ROS and mitochondria. However, despite the physiological evidence of the involvement of oxidative stress in multiple pathologies, such as Alzheimer’s, cancers or diabetes, the mode of action of ROS remains incompletely elucidated.
Reticulum stress
The reticulum is an organelle that allows the synthesis, modification and folding of proteins. There are two types of reticulum: rough, which has ribosomes and translates RNA into proteins, and smooth, which allows post-translational modifications. It is an essential element for the good function of our cells and one of the favourite targets of cellular stress.
Calcium deregulation
The production of proteins which takes place in the reticulum is an oxidative mechanism and requires a specific environment allowing the formation of structures in proteins (disulfide bridges, glycosylation…), essential for their folding. It is also very rich in calcium and chaperon proteins[10]. Because of this oxidative environment, the reticulum releases ROS which, when proteins fold poorly or the cell is subjected to stress, are produced in excess. They can then settle on membrane channels. These structures are, as their name suggests, small tunnels through which all kinds of molecules can pass, starting with ions, such as calcium (Ca2+). The reticulum will therefore, following the regulation of its calcium channels by the ROS, release Ca2+ into the cytoplasm, trigger stress signals and generate a cellular response[11]. One of these signals consists in sending Ca2+ into the mitochondria which, when the calcium concentration increases, will produce new ROS and maintain a self-activating loop of cellular stress.
In addition to this, the calcium overflow will lead to the activation of synthesis pathways leading to the production of pro-inflammatory cytokines (such as NFκB) and chaperone proteins (in order to cope with the abundance of poorly folded proteins): this is called the “overload reticulum response”[12].
Poor protein folding
To complete the vicious circle of cellular stress, when the reticulum is stressed, it produces a lot of ROS and releases calcium, thus activating pro-stress signalling pathways, it also maintains its own stress by producing more and more poorly folded proteins. This triggers UPR (“Unfolded Protein Response”), a mechanism developed in the first part of this dossier, which will force the cell to choose between survival pathway or activation of apoptosis.
Thus, the reticulum can respond to cellular stress in several ways[13]
- attenuation of transcriptional response through gene regulation to reduce the number of proteins produced,
- upward regulation of genes encoding chaperone proteins, via calcium regulation, to correctly fold proteins that are not chaperone proteins,
- synthesis of pro-inflammatory cytokines via calcium regulation,
- the implementation of apoptosis, when the first three options have failed.
Nucleus stress and genotoxic mechanisms
Gene regulation is essential to the response to cellular stress and involves various mechanisms, including reticulum and mitochondrial stress. It allows, indeed, an adaptation of the proteins produced and an adjustment of the synthesized enzymes, in order to answer the specific requests of the cell at the time of the aggression. However, there is another type of stress that comes from the core itself. DNA is a target like any other when stress is involved, and many chemicals or external events can cause damage to its structure. DNA is very sensitive to chemotherapeutic treatments, irradiation or genotoxic elements such as UV. It is most fragile when it is being transcribed or replicated, normal cellular steps during which it occurs in its single-stranded form and where it can be broken. Fortunately, there is a whole machinery allowing its repair: the break is recognized by specific enzymes (ERCC1-XPF and XPG) which can then act and repair small single-stranded lesions[14]. However, when the break occurs on the double strand of DNA, a completely different response takes place: enzymes are recruited (notably the MRN complex) to trigger a central signalling pathway, that of p53[15, 16]. p53 is an important protein in the regulation of apoptosis and its activation will trigger the synthesis of pro-apoptotic factors, but also stimulate mitochondria and promote cell death.
Peroxysome proliferation

Ever heard of peroxysomes? This is normal, there is little discussion about them, although their role is important for our cells because they participate in their detoxification, via the degradation of certain fatty acids and alcohols. One downside, when they degrade these elements, they produce H2O2 . To some extent, catalases present in the peroxysome can convert H2O2 to H2O and O2, thus allowing recycling of this toxic molecule, while reinjecting oxygen into cellular metabolism[17]. On paper, it’s beautiful. However, we do not understand very well how peroxysomes maintain a balance between produced and degraded ROS. Moreover, in case of peroxisome problems, H2O2 is largely released into the cytoplasm significantly increasing oxidative cellular stress[17].
Peroxysomes can also multiply via the action of proliferation activators on their nuclear receptors, PPARs (Peroxysome Proliferation Activator Receptors). PPARs can be activated by different environmental, nutritional or inflammatory signals, such as prostaglandins, certain fatty acids or pro-inflammatory leukotriene, arachidonic acid, certain hypolipidemic agents and NSAIDs. There are three forms of PPARs, α, β and ϒ, which are very similar, but whose tissue expression can vary. When activated, they create heterodimers with another receptor capable of binding to DNA and thus regulate the expression of certain proteins, whose gene contains a promoter with a PPAR (peroxisome proliferator response element) recognition sequence[18]. Among these genes, we obviously find those involved in the proliferation of peroxysomes, but also certain genes coding for mitochondrial respiratory chain proteins or antioxidants.
All these organelles affected by cellular stress are deeply modified and determine the survival of the cell as a whole. During severe stress, it is not uncommon to observe the death of thousands of cells in parallel that can have an impact on our health. During aging, these processes are more and more frequent and partially explain why our body feels its age!
A few definitions
Caspases : Caspases are proteins that play a central role in programmed cell death (mostly apoptosis) and inflammation. They are enzymes capable of activating many signaling pathways via the cysteine protease activity of their active site.
Membrane potential : cell membranes are phospholipid bilayers, i.e. they are formed by two layers of modified lipids. There is therefore an internal membrane, an inter-membrane space and an external membrane. Ions (positive or negative) are present inside and outside these different membranes. In order to prevent them from entering and leaving as they wish and deregulate cellular homeostasis (maintenance of pH, enzyme function, protein synthesis, etc.), membranes are impermeable and manage the entry and exit of these ions, thus creating a membrane potential, i.e. the difference between the potential inside the inner membrane and the potential outside the outer membrane.
NSAIDs : Non-steroidal anti-inflammatory drugs, such as aspirin or ibuprofen.
All our articles on Cellular stress and aging:
Cellular stress and aging, a necessary duo?
 Although it is recognized as a risk factor by the scientific community, many questions remain regarding cellular stress. Its main role in aging-related diseases and in aging itself is still being studied.
Although it is recognized as a risk factor by the scientific community, many questions remain regarding cellular stress. Its main role in aging-related diseases and in aging itself is still being studied.
Part 1: The different types of cellular stress
 Cellular stress can mean many different mechanisms, whether the stress is mechanical, toxic, chemical, thermic, osmotic, ionizing… They all play a role in the aging process of our cells.
Cellular stress can mean many different mechanisms, whether the stress is mechanical, toxic, chemical, thermic, osmotic, ionizing… They all play a role in the aging process of our cells.
Part 2: The organelles targeted by cellular stress
 Cellular stress gets transmitted through several organelles in our cells, especially those of the mitochondria, the mitochondria and the nucleus, among others. Their response can vary and only give the cell under stress two viable options: adapt and survive, or die.
Cellular stress gets transmitted through several organelles in our cells, especially those of the mitochondria, the mitochondria and the nucleus, among others. Their response can vary and only give the cell under stress two viable options: adapt and survive, or die.
Part 3: Dealing with cellular stress
 In order to slow down the aging process of our cells, we must control our cellular stress. In this article, we will look into the techniques and/or treatments currently used to treat all forms of cellular stress.
In order to slow down the aging process of our cells, we must control our cellular stress. In this article, we will look into the techniques and/or treatments currently used to treat all forms of cellular stress.
References
[1] Friedman JR & Nunnari J, Mitochondrial form and function, Nature, 2014;505:335–343
[2] Turrens JF, Boveris A, Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 1980;191:421–427
[3] Matés JA, Segura FJ, Alonso JM, Javier M, Oxidative stress in apoptosis and cancer: an update. Arch Toxicol. 2012;86:11, pp 1649–1665
[4] Masutani H, Yodoi J (2002) Thioredoxin. Overview. Methods Enzymol 347:279–286
[5] Farrugia G, Balzan R, Oxidative stress and programmed cell death in yeast. Front Oncol 2012;2:64
[6] Gabai VL, Yaglom JA, Volloch V et al., Hsp72-mediated suppression of c-Jun N-terminal kinase is implicated in development of tolerance to caspase-independent cell death. Mol Cell Biol 2000;20:6826–6836
[7] Barnhart BC, Alappat EC, Peter ME, The CD95 type I/type II model. Semin Immunol 2003;15:185–193
[8] Circu ML, Aw TY, Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 2010;48:749–762
[9] Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
[10] Schröder M, Kaufman RJ, ER stress and the unfolded protein response. Mutat Res. 2005 Jan 6; 569(1-2):29-63
[11] Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. Journal of Clinical Investigation. 2005;115(10):2656-2664
[12] Kong L, Li S, Huang M, et al. The Roles of Endoplasmic Reticulum Overload Response Induced by HCV and NS4B Protein in Human Hepatocyte Viability and Virus Replication. Shoukry NH, ed. PLoS ONE. 2015;10(4):e0123190
[13] Oyadomari S, Mori M, Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell death and differentiation. 2004, 11. 381-9
[14] L. Staresincic, A. F. Fagbemi, J. H. Enzlin et al., Coordination of dual incision and repair synthesis in human nucleotide excision repair, EMBO Journal, 2009, vol. 28, no. 8, pp. 1111–1120
[15] M. Christmann, M. T. Tomicic, W. P. Roos, and B. Kaina, Mechanisms of human DNA repair: an update, Toxicology, 2003, vol. 193, no. 1-2, pp. 3–34
[16] J. W. Harper and S. J. Elledge, The DNA damage response: ten years after, Molecular Cell, 2007 vol. 28, no. 5, pp. 739–745
[17] Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J, Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem 2004;266:37–56
Dr. Marion Tible
Author/Reviewer
Auteure/Relectrice
Marion Tible has a PhD in cellular biology and physiopathology. Formerly a researcher in thematics varying from cardiology to neurodegenerative diseases, she is now part of Long Long Life team and is involved in scientific writing and anti-aging research.
More about the Long Long Life team
Marion Tible est docteur en biologie cellulaire et physiopathologie. Ancienne chercheuse dans des thématiques oscillant de la cardiologie aux maladies neurodégénératives, elle est aujourd’hui impliquée au sein de Long Long Life pour la rédaction scientifique et la recherche contre le vieillissement.
En savoir plus sur l’équipe de Long Long Life
Dr Guilhem Velvé Casquillas

Author/Reviewer
Auteur/Relecteur
Physics PhD, CEO NBIC Valley, CEO Long Long Life, CEO Elvesys Microfluidic Innovation Center
More about the Long Long Life team
Docteur en physique, CEO NBIC Valley, CEO Long Long Life, CEO Elvesys Microfluidic Innovation Center
En savoir plus sur l’équipe de Long Long Life











