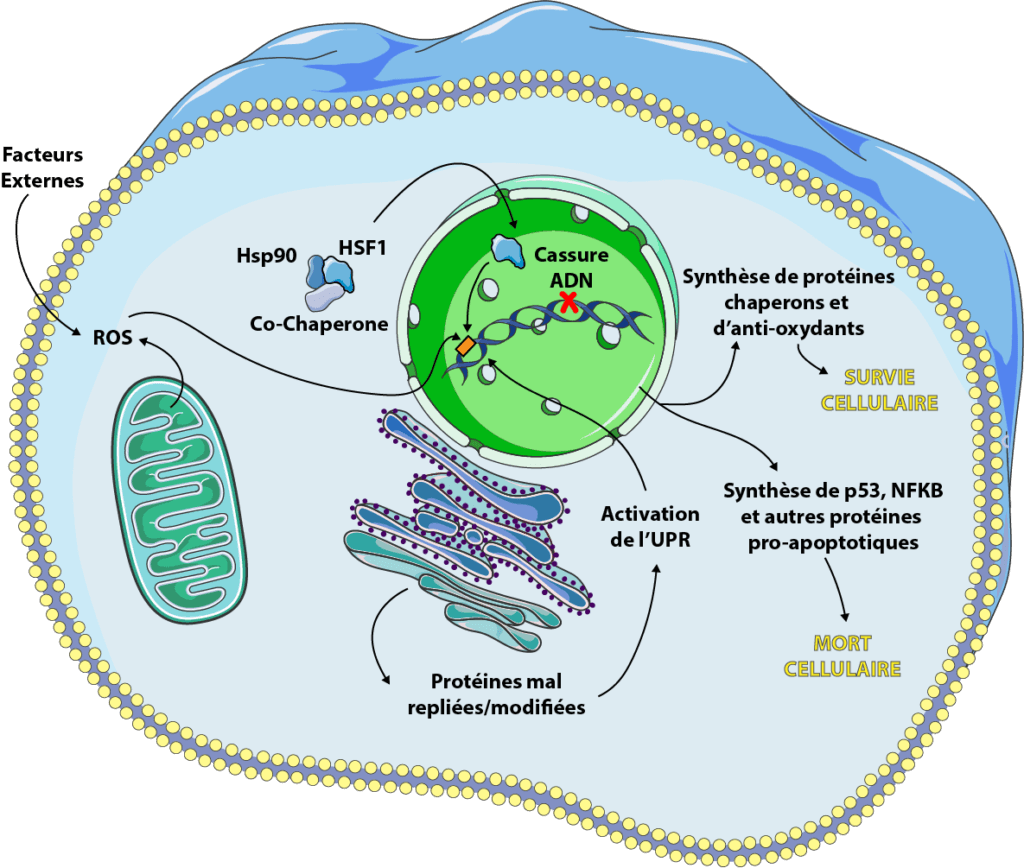
Fight Aging! Extraits
Parce que tout le monde ne comprend pas forcément la langue de Shakespeare, Long Long Life vous fournit une traduction automatique post-éditée des éléments les plus importants de la newsletter Fight Aging!
Fight Aging! fournit un résumé hebdomadaire des actualités et des commentaires pour des milliers d’abonnés intéressés par la science de la longévité: progrès en matière de contrôle médical du vieillissement afin de prévenir la vulnérabilité, la souffrance et les maladies liées à l’âge, ainsi que pour fournir des améliorations dans la compréhension actuelle de ce qui fonctionne et ce qui ne fonctionne pas lorsqu’il s’agit de prolonger une vie saine. Attendez-vous à voir des résumés des récents progrès de la recherche médicale, des nouvelles de la communauté scientifique, des initiatives de collecte de fonds pour accélérer le travail sur la réparation et l’inversion du vieillissement, des liens vers des ressources en ligne, et bien plus encore.
Ce contenu est publié sous la licence Creative Commons Paternité 4.0 International License. Nous vous encourageons à le republier et à le réécrire comme bon vous semble, la seule condition étant de fournir une attribution et un lien vers Fight Aging!
Pour vous abonner ou vous désabonner, veuillez visiter: https://www.fightaging.org/newsletter/
Extraits de Fight Aging! :
- Nouvelles découvertes sur l’aspect biochimique de la maladie de Parkinson
- Une thérapie génique pour réduire considérablement l’incidence des maladies cardiovasculaires
- Financer des travaux supplémentaires sur le Deep Learning pour découvrir des médicaments contre le vieillissement
- Les médicaments sénolytiques ne parviennent pas à tuer les cellules cancéreuses avec des signatures d’expression de gènes sénescents, mais une thérapie génique réussit
- L’aspirine en tant que mimétique de restriction calorique améliorant l’autophagie
Nouvelles découvertes sur l’aspect biochimique de la maladie de Parkinson
Aujourd’hui, je souligne quelques résultats de recherche publiés récemment qui contribuent à la compréhension de la maladie de Parkinson et de sa progression. La maladie de Parkinson est relativement simple, comme les maladies neurodégénératives – c’est-à-dire que sa biochimie est encore extrêmement complexe en détail, mais il n’est pas aussi difficile d’identifier les aspects importants que la maladie d’Alzheimer. À la racine, il s’agit d’une synucléinopathie, une condition causée par l’accumulation de dépôts d’α-synucléine. Il en résulte un dysfonctionnement mitochondrial et la mort cellulaire d’une petite mais importante population de neurones dopaminergiques reliés à la fonction motrice, mais aussi une perturbation plus généralisée de la fonction normale du cerveau. Le défi dans la maladie de Parkinson est moins de savoir où intervenir, c’est-à-dire l’élimination ciblée de la synucléine, que de construire une méthodologie efficace. Vous pouvez consulter l’une des revues de la Fondation de recherche SENS sur le sujet pour vous faire une idée de la difficulté d’éliminer en toute sécurité une forme spécifique de déchets métaboliques du cerveau.
Pourquoi certaines personnes seulement développent la maladie de Parkinson? Dans un petit nombre de cas, c’est dû à des gènes mutants, en particulier ceux comme la parkine qui sont importants dans les processus d’entretien cellulaire. La déficience de l’autophagie axée sur le contrôle de la qualité des mitochondries semble être une facette importante de la maladie de Parkinson, mais chez les patients qui n’ont pas de signes de mutation, le cheminement vers la maladie peut être aléatoire, c’est-à-dire de petites différences dans les dommages liés à l’âge et la diminution de l’entretien cellulaire qui s’accumulent et s’accélèrent avec le temps. Un peu moins d’élimination des déchets métaboliques conduit à un peu plus de synucléine? et un peu plus de dysfonctionnement mitochondrial, ce qui à son tour a un impact sur les systèmes de maintenance, et donc la boucle de rétroaction progresse, toujours plus vite au fil du temps. Avec suffisamment de temps, tout le monde finira par souffrir de la maladie de Parkinson, mais à l’heure actuelle, d’autres processus de vieillissement tuent la plupart des gens avant que cela ne se produise.
Au-delà de la clairance de la synucléine, la thérapie cellulaire est l’autre domaine d’activité majeur dans la production de thérapies pour la maladie de Parkinson. L’objectif est de remplacer les neurones dopaminergiques perdus par de nouvelles cellules capables de reprendre la même fonction dans le cerveau. Comme le processus de perte est graduel, cela devrait apporter un bénéfice durable, même sans s’attaquer aux causes de la perte cellulaire – les nouvelles cellules seront détruites à temps, tout comme les anciennes. Cette situation est semblable dans presque tous les cas d’utilisation proposée de la thérapie de remplacement cellulaire chez les personnes âgées: l’environnement tissulaire est généralement hostile et endommagé, et les détails sur la façon dont et pourquoi les populations cellulaires existantes ne permettent plus un bon fonctionnement lorsqu’il s’agit de l’efficacité potentielle de l’introduction de nouvelles cellules. Vont-elles fonctionner correctement ou succomber rapidement?
Des chercheurs découvrent un coupable de la mort des cellules cérébrales de Parkinson
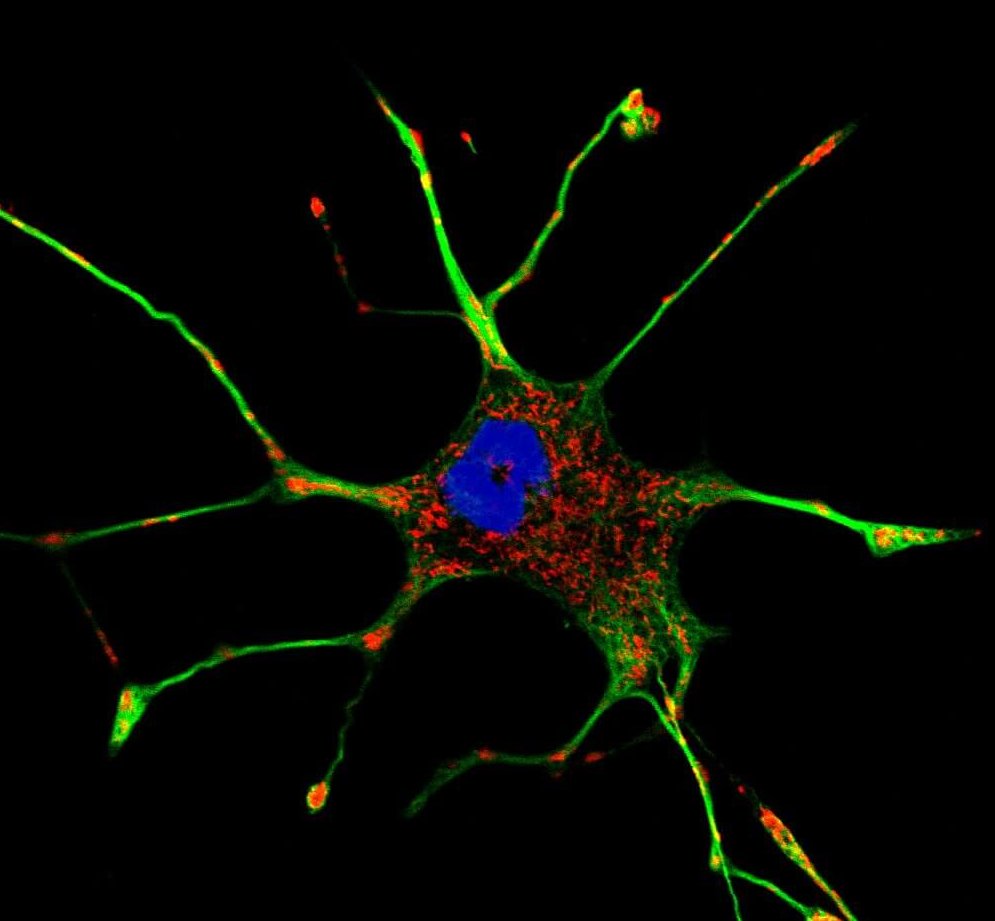
Si nous pouvions regarder dans le cerveau des patients atteints de la maladie de Parkinson, nous verrions deux signes distinctifs de la maladie. D’abord, on verrait mourir les cellules cérébrales qui produisent une substance chimique appelée dopamine. On verrait aussi des grappes de protéines appelées corps de Lewy dans les neurones. Les chercheurs croient qu’une des clés du traitement de la maladie de Parkinson est d’étudier les liens possibles entre ces deux phénomènes. « Cette étude identifie le lien manquant entre les corps de Lewy et le type de dommages observés dans les neurones touchés par la maladie de Parkinson. La maladie de Parkinson est un trouble des mitochondries, et nous avons découvert comment les corps de Lewy libèrent un produit de décomposition partielle qui a un tropisme élevé pour les mitochondries et détruit leur capacité à produire de l’énergie. »
Les corps de Lewy ont été décrits il y a un siècle, mais ce n’est qu’en 1997 que les scientifiques ont découvert qu’ils étaient faits de touffes d’une protéine mal pliée appelée synucléine. Lorsqu’elle n’est pas mal pliée, on croit que la synucléine exerce des fonctions liées à la transmission de signaux entre les neurones. Les chercheurs se sont penchés sur les cultures cellulaires de neurones qui ont été incités à accumuler des fibrilles constituées de synucléine-α mal pliée, imitant le corps de Lewy chez les patients atteints de la maladie de Parkinson. Ils ont découvert que lorsque les fibrilles d’α-synucléine sont décomposées, elles créent souvent un amas protéique plus petit, qu’ils appellent pα -syn*.
Il s’avère que le résultat de cette dégradation partielle, pα -syn*, est toxique. Les chercheurs ont fait cette découverte en étiquetant le pα -syn* avec un anticorps afin qu’ils puissent le suivre dans toute la cellule après sa création. Ils ont observé que pα -syn* voyageait et s’attachait aux mitochondries. Une enquête plus poussée a révélé qu’une fois la pα -syn* attachée, les mitochondries ont commencé à se dégrader. Ces mitochondries fragmentées perdent leur capacité à transmettre un signal électrochimique et à produire de l’énergie. « Les corps de Lewy sont de gros agrégats et ils restent dans la cellule, mais ils n’entrent pas en contact direct avec les mitochondries comme le fait pα -syn*. Avec cette découverte, nous avons établi un lien direct entre la protéine synucléine et les effets en aval qui sont observés lorsque les cellules cérébrales sont endommagées dans la maladie de Parkinson. »
Une étude révèle de nouvelles connaissances sur la cause de la mort cellulaire dans la maladie de Parkinson
Les chercheurs ont découvert que la cardiolipine, une molécule à l’intérieur des cellules nerveuses, aide à s’assurer qu’une protéine appelée alpha-synucléine se plie correctement. Un mauvais pliage de cette protéine entraîne des dépôts de protéines qui sont la marque distinctive de la maladie de Parkinson. Ces dépôts sont toxiques pour les cellules nerveuses qui contrôlent les mouvements volontaires. Lorsque trop de ces dépôts s’accumulent, les cellules nerveuses meurent. « L’identification du rôle crucial que joue la cardiolipine dans le maintien de la fonctionnalité de ces protéines signifie que la cardiolipine peut représenter une nouvelle cible pour le développement de traitements contre la maladie de Parkinson. »
L’étude a révélé qu’ à l’intérieur des cellules, l’alpha-synucléine se lie aux mitochondries, où réside la cardiolipine. Les cellules utilisent les mitochondries pour produire de l’énergie et stimuler le métabolisme. Normalement, la cardiolipine présente dans les mitochondries extrait la synucleine des dépôts de protéines toxiques et la replie en une forme non toxique. Les chercheurs ont découvert que chez les personnes atteintes de la maladie de Parkinson, ce processus est dépassé au fil du temps et que les mitochondries sont détruites. La compréhension du rôle de la cardiolipine dans le repliement des protéines peut aider à créer un médicament ou une thérapie pour ralentir la progression de la maladie.
Une thérapie génique pour réduire considérablement l’incidence des maladies cardiovasculaires
Il semble plausible que l’un des premiers grands axes de développement de la thérapie génique chez l’humain sera celui des gènes invalidants qui maintiennent les taux de lipides dans la circulation sanguine. Il existe un certain nombre de cibles crédibles: PCSK9, ANGPTL3, ANGPTL4 et ASGR1 par exemple. La réduction des taux de lipides dans la circulation sanguine a pour effet de ralentir le développement des maladies cardiovasculaires, ce qui réduit le risque de crise cardiaque, d’accident vasculaire cérébral et d’autres problèmes connexes. Le succès des statines repose précisément sur cet effet, et les thérapies géniques seraient beaucoup plus efficaces que les statines – un traitement unique produisant un bénéfice plus important et permanent.
Comment ça marche sous le capot? Pourquoi l’abaissement des lipides sanguins – cholestérol, triglycérides, et ainsi de suite – a-t-il ce résultat bénéfique? Le principal mécanisme d’intérêt concerne le développement de l’athérosclérose par des molécules lipidiques endommagées. Le fonctionnement normal du métabolisme produit des molécules réactives qui peuvent oxyder les lipides, de sorte qu’une petite fraction des lipides dans la circulation sanguine sont endommagés de cette façon. Avec la progression du vieillissement, diverses formes de dommages aux cellules et aux tissus et leurs conséquences conduisent à la génération de molécules beaucoup plus réactives, et donc à un plus grand nombre de molécules lipidiques endommagées entrant dans la circulation sanguine – le problème s’aggrave avec le temps. Vous pourriez examiner la progression de la cause et de l’effet qui commence avec les dommages mitochondriaux à l’ADN, par exemple, mais il y a aussi des problèmes plus systémiques comme l’inflammation chronique, qui va de pair avec des niveaux plus élevés de dommages oxydatifs.
Les lipides oxydés peuvent irriter les parois des vaisseaux sanguins, provoquant une boucle de rétroaction de réactions cellulaires inappropriées qui produisent l’inflammation et attirent de plus en plus de cellules immunitaires pour essayer de nettoyer le désordre. Beaucoup de ces cellules meurent, submergées par des formes de lipides oxydés que la biochimie mammalienne n’est pas bien équipée pour manipuler. Il en résulte une plaque de graisse croissante de cellules mortes et des lipides nocifs, signature de l’athérosclérose. Ces plaques affaiblissent et rétrécissent les vaisseaux sanguins, et finalement quelque chose se rompt ou un vaisseau sanguin est bloqué – un événement souvent fatal, et au mieux invalidant. Interférer dans cette boucle de rétroaction à n’importe quel point peut la ralentir: réduire la quantité de tous les lipides entrant dans la circulation sanguine, rendre les cellules immunitaires plus résilientes ou capables, ou enlever seulement les lipides problématiques via un autre mécanisme.
Je pense que ces deux dernières sont meilleures que les premières, car elles peuvent en principe être rendues efficaces à près de 100 % sans altérer le fonctionnement du métabolisme cellulaire de manière à ce qu’il reste à comprendre à long terme, comme c’est le cas pour les réductions spectaculaires des taux de lipides sanguins. Ce n’est pas le cas que tous les lipides peuvent être retirés de la circulation sanguine, et ce n’est pas le cas que tous les dommages oxydatifs peuvent être évités. En général, une réparation périodique est bien plus utile qu’une prévention partielle, car la réparation peut aider les personnes déjà endommagées et aux stades avancés de la maladie. Néanmoins, nous obtiendrons quand même une mise en œuvre plus efficace de la pire option dans un avenir proche, semble-t-il, car c’est là que l’attention se concentre le plus.
Un traitement CRISPR pour les maladies cardiaques
Considérez ce scénario: nous sommes en 2037, et une personne d’âge moyen peut se rendre dans un centre de santé pour se faire vacciner contre les maladies cardiovasculaires. L’injection cible les cellules du foie, en modifiant un gène qui intervient dans la régulation du cholestérol sanguin. La procédure simple réduit le taux de cholestérol et réduit considérablement le risque de crise cardiaque. Bien que des thérapies à base d’anticorps aient été lancées pour aider les personnes les plus à risque, le coût et la complexité des traitements signifient qu’une solution plus simple et unique, comme un vaccin, serait bénéfique pour de nombreuses autres personnes dans le monde.
La bonne nouvelle, c’est qu’une combinaison de la découverte de gènes et de l’éclosion de technologies d’édition de génomes telles que CRISPR-Cas9 a donné à cette vision d’un avenir fondé sur le vaccin pour lutter contre les maladies cardiaques une forte chance de devenir réalité. La percée a eu lieu en 2003, lorsque les chercheurs ont enquêté sur trois familles françaises dont les membres présentaient des taux potentiellement mortels de cholestérol LDL (Low-density lipoprotein) et qui présentaient une mutation du gène PCSK9. PCSK9 code une enzyme qui régule les niveaux de LDL – ou « mauvais » cholestérol. Décelant les possibilités, les chercheurs ont cherché à déterminer si des mutations naturelles dans le PCSK9 pourraient également avoir pour effet d’abaisser le cholestérol LDL. Après avoir passé en revue les données d’environ 3 600 personnes ayant fourni un échantillon de sang, les chercheurs ont séquencé l’ADN des 128 participants ayant les plus faibles taux de cholestérol LDL. Ils ont découvert qu’environ 2% des participants afro-américains avaient une copie cassée du PCSK9. Une étude de suivi d’une population différente et plus nombreuse a également révélé des mutations chez près de 3 % des Afro-Américains, ce qui a été associé à une réduction de 88 % du risque de cardiopathie ischémique.
Le foie est un organe cible privilégié de la thérapie génique pour des sociétés telles que Editas Medicine, Sangamo Therapeutics et CRISPR Therapeutics; il est facile de délivrer des gènes au foie, et l’outil CRISPR-Cas9 est particulièrement efficace dans l’organe, éditant une plus grande proportion de cellules que dans la plupart des autres tissus. Le foie est également un excellent endroit pour lutter contre le cholestérol – il libère le cholestérol LDL du sang et est également un moteur principal de la synthèse lipidique. Des chercheurs ont montré que plus de la moitié des gènes Pcsk9 présents dans le foie de la souris pourraient être réduits au silence par une seule injection d’un adénovirus contenant un système CRISPR-Cas9 dirigé contre Pcsk9. Cela a entraîné une diminution d’environ 90 % du taux de Pcsk9 dans le sang et une baisse de 35 à 40 % du taux de cholestérol LDL dans le sang.
L’approche est « absolument plausible, voire réalisable » d’un point de vue technologique. Mais il y a aussi une barrière philosophique à négocier. « Vous ne voulez pas nécessairement soigner des gens qui n’ont pas encore de maladie. » D’autres vont plus loin. « Changer de mode de vie peut être beaucoup plus efficace pour une population que de se concentrer sur des interventions coûteuses. » Ils craignent qu’une thérapie génique pour les personnes à haut risque n’entrave les efforts pour aider les gens à s’aider eux-mêmes. « C’est la façon dont l’esprit humain travaille. Prenez une pilule et nous pensons être protégés. »
Il y a certainement une certaine réticence à poursuivre les thérapies géniques permanentes pour la prévention et l’amélioration en ce moment – les travaux pourraient avancer beaucoup plus rapidement qu’ils ne le font actuellement, étant donné la baisse rapide des coûts de la biotechnologie génétique. Il faudra probablement des groupes plus aventureux comme BioViva Sciences ou Ascendance Biomédical pour briser cette porte en allant tout simplement de l’avant et en offrant les thérapies géniques qui sont technologiquement plausibles en dehors du système réglementaire principal. Ces traitements auront d’abord une faible efficacité, en termes de proportion de cellules transfectées par la thérapie, mais c’est un défi qui sera résolu avec une efficacité croissante au cours de la prochaine décennie. Quelqu’un doit commencer, être le premier, montrer le chemin. Si les systèmes de réglementation tels qu’ils existent actuellement rendent cela difficile, alors le début se produira en dehors du cadre réglementaire, tout comme il l’ a fait pour les thérapies à base de cellules souches – et c’est une bonne chose aussi, car c’est pratiquement la seule circonstance qui pourrait aider à rendre la réglementation médicale actuelle moins oppressante.

Financer des travaux supplémentaires sur le Deep Learning pour découvrir des médicaments contre le vieillissement
Récemment, Y Combinator a annoncé son intention de financer des entreprises qui travaillent sur les traitements pour le vieillissement. C’est l’un des nombreux signes d’un intérêt croissant des investisseurs pour ce domaine du développement. L’un des premiers résultats semble être un plus grand financement pour les méthodes informatiques d’amélioration de la découverte de médicaments, avec des thérapies pour le vieillissement comme but commun, après le modèle établi d’Insilico Médecine. Il est logique qu’une partie de cette communauté principalement axée sur le logiciel se lancerait dans un nouveau domaine, la biotechnologie, en finançant des entreprises qui appliqueraient la technologie informatique à ce domaine. Cela ne dit rien sur l’efficacité de la démarche, bien sûr, mais il s’agit d’une évolution naturelle des connaissances et des intérêts établis.
Il y a certainement beaucoup de place pour l’amélioration en ce qui concerne le coût et l’effort requis pour trouver et prouver de petites molécules et d’autres médicaments pour traiter des maladies spécifiques ou cibler des mécanismes biologiques spécifiques avec un minimum d’effets secondaires. Il est raisonnable de penser que les approches deep learning établies peuvent être appliquées avec succès ici, pour attirer l’attention sur les molécules des bibliothèques standard qui pourraient autrement être négligées, et pour concevoir de nouvelles molécules thérapeutiques basées sur les données existantes et les caractéristiques souhaitées. Il y a cependant une différence importante entre, disons, l’application de cette technologie à la recherche de sénolytiques et de disjoncteurs de liaison croisée, des approches qui peuvent en principe produire un rajeunissement, ou l’application à la recherche d’autres géroprotecteurs comme la metformine, la rapamycine, etc. Ces derniers ne peuvent ralentir que marginalement la progression du vieillissement, et le milieu de la recherche peine à produire quelque chose dans cette partie du domaine qui puisse faire mieux que l’exercice et la restriction calorique. Il reste à voir dans quelle direction.
Au cours des dernières décennies, un nombre incommensurable de preuves se sont accumulées : il est possible de ralentir les processus biologiques du vieillissement chez les animaux. Ces données probantes s’accumulent le long de multiples axes de recherche couvrant de nombreuses thérapies différentes. Nous en sommes arrivés à la même conclusion: en comprenant et en traitant directement les dommages biologiques accumulés pendant le vieillissement, nous pouvons trouver de nouvelles thérapies puissantes pour lutter contre la maladie et vivre plus longtemps et en meilleure santé.
Chez Spring Discovery, nous accélérons la découverte de ces thérapies grâce à notre plateforme de découverte de médicaments basée sur le deep learning. Et nous sommes fiers d’annoncer que nous avons récolté un financement de 4,25 millions de dollars auprès d’une équipe de bailleurs de fonds en biotechnologie qui appuient notre vision à long terme. Pourquoi les thérapies axées sur le vieillissement représentent-elles une opportunité si considérable? Parce que le vieillissement est le seul facteur de risque le plus important pour les maladies les plus nuisibles sur Terre – les maladies cardiovasculaires, les maladies neurodégénératives, les maladies pulmonaires, le cancer, la perte musculaire et plus encore – et les médicaments qui ralentissent les dommages biologiques accumulés pendant le vieillissement ont le potentiel de réduire l’incidence de ces maladies, peut-être simultanément.
Ensemble, le vieillissement représente A) l’un des problèmes les plus importants auxquels l’humanité est confrontée et B) un problème qui semble de plus en plus possible à résoudre. Les maladies de la vieillesse ne sont pas discriminatoires, mais elles peuvent être combattues. Nous croyons que dans un avenir pas trop lointain, la découverte de thérapies pour le vieillissement fournira certains des outils les plus efficaces de l’histoire pour réduire notre fardeau de la maladie et prolonger notre vie en santé. Spring Discovery a pour mission d’accélérer considérablement la réalisation de cet avenir. Et nous apportons un nouvel ensemble d’outils d’apprentissage machine pour relever ce défi.
Les médicaments sénolytiques ne parviennent pas à tuer les cellules cancéreuses avec des signatures d’expression de gènes sénescents, mais une thérapie génique réussit
Certaines cellules cancéreuses expriment des signatures normalement associées aux cellules sénescentes, alors pourquoi ne pas essayer des composés sénolytiques contre elles? Il s’agit là d’un cercle vicieux, étant donné que la plupart des médicaments candidats sénolytiques actuels ont été à l’origine caractérisés et testés comme chimiothérapeutiques potentiels. Ce document en libre accès est intéressant pour deux raisons: premièrement, le fait que les médicaments sénolytiques n’ont pas tué les cellules cancéreuses portant une signature sénescente et, deuxièmement, qu’une thérapie génique par suicide ciblant cette signature agit contre les cellules sénescentes normales et les cellules cancéreuses portant une signature sénescente. L’approche de la thérapie génique décrite ici est conceptuellement similaire (à un niveau très élevé) à la thérapie génique de Biotechnologies Oisin utilisée pour détruire les cellules sénescentes, mais moins flexible. Les fondateurs d’Oisin Biotechnologies ont montré que le ciblage de p53, un suppresseur de cancer, plutôt que p16 / p16Ink4a, une signature de sénescence, est très efficace contre le cancer, mais il semble que p16 est également un déclencheur viable pour les mécanismes de thérapie génique de destruction cellulaire dans de nombreux cancers.
p16Ink4a arrête la progression du cycle cellulaire en inhibant la phase S. La sénescence cellulaire, un mécanisme suppressif de tumeur défini comme arrêt de croissance irréversible et induit par l’accumulation de dommages d’ADN, est souvent associée à l’induction de p16Ink4a. Par conséquent, p16Ink4a est considéré comme un puissant suppresseur de tumeur. Les mutations de perte de fonction affectant p16Ink4a sont une marque commune de diverses tumeurs humaines, et sont considérées comme une étape essentielle vers la progression tumorale. Cependant, en présence de mutations affectant RB ou CDK4/CDK6, l’activité p16Ink4a n’est pas suffisante pour arrêter la progression du cycle cellulaire. De plus, une surexpression de p16Ink4a a été observée sur le front envahissant du carcinome endométrial, colorectal et basocellulaire et corrélée à une forte agressivité. Ainsi, dans ces conditions, cibler les cellules surexprimant p16Ink4a pourrait constituer une intervention anticancéreuse puissante.
Malgré le pontage par mutation du programme de sénescence, les cellules de sarcome surexprimant le SAR et avec p53 inactif ont induit un niveau élevé de p16Ink4a. Nous avons alors émis l’hypothèse que le traitement avec des composés dont on a montré qu’ils éliminent sélectivement les cellules surexprimant la p16Ink4a sénescente pourrait être une stratégie efficace. Deux des composés les plus efficaces aux propriétés sénolytiques (c’est-à-dire sélectivement toxiques contre les cellules sénescentes) sont l’ABT-263 et l’ABT-737, des agents anticancéreux bien connus inhibant la famille des protéines antiapoptotiques BCL2. Cependant, aucun des deux traitements n’était toxique pour ces cellules cancéreuses. Cela suggère que les cellules tumorales surexprimant p16Ink4a sont résistantes aux composés actuellement disponibles avec une spécificité contre les cellules p16Ink4a+.
Nous avons alors estimé qu’une stratégie alternative pour l’élimination des cellules tumorales surexprimant p16Ink4a pourrait faire appel à la thérapie génique ciblée. La thérapie génique du suicide a été étudiée dans divers types de cancer en raison de sa spécificité supérieure à celle des thérapies génotoxiques standard. Un effort antérieur pour tester une thérapie génique suicidaire sous la réglementation du promoteur p16Ink4a – le système INK-ATTAC – n’ a pas réussi à tuer les cellules cancéreuses p16Ink4a+, bien qu’il ait été efficace pour éliminer les cellules sénescentes p16Ink4a+. Nous avons récemment développé un système de suicide similaire, appelé p16-3MR. La principale différence réside dans le fait que le gène p16-3MR est soumis à la régulation du promoteur p16Ink4a complet, tandis que l’INK-ATTAC est régulé par une petite partie à proximité du site de transcription du locus INK4a.
Notre stratégie, qui s’est révélée très efficace dans les cellules non propagatrices, a montré une toxicité élevée pour les cellules cancéreuses, tant en culture cellulaire qu’in vivo. De plus, puisqu’il a été démontré que dans certains cas, les cellules p16Ink4a+ sont des précurseurs de cellules malignes, le système 3MR pourrait permettre de réduire l’incidence des tumeurs en éliminant les cellules précancéreuses p16Ink4a+. A ce stade, des recherches approfondies devraient être menées pour tester la toxicité d’une stratégie de thérapie génique suicidaire pilotée par p16Ink4a contre d’autres types de tumeurs.

L’aspirine en tant que mimétique de restriction calorique améliorant l’autophagie
L’une des meilleures façons d’atténuer le battage publicitaire inutile généré par tel ou tel nouveau supplément ou médicament qui ralentirait modestement le vieillissement selon les données animales est de souligner que l’aspirine fait tout aussi bien l’affaire dans les études animales. Nous savons tous ce que l’aspirine fait pour la durée de vie humaine, c’est-à-dire à peu près rien, tout en continuant d’être un outil utile dans la boîte à outils pharmaceutique. La poursuite de résultats marginaux dans la longévité humaine permettra au mieux d’atteindre des résultats marginaux – et c’est le principal problème avec l’accent sur la tendance générale à essayer de récupérer les effets bénéfiques de la restriction calorique par le biais d’un certain nombre de candidats à la restriction calorique des médicaments mimétiques. Nous devons faire mieux, viser plus haut. Cela signifie qu’il faut davantage de travaux axés sur la mise au point de thérapies selon le modèle SENS, c’est-à-dire celles qui réparent les dommages moléculaires qui causent le vieillissement et qui sont donc capables, en principe, de rajeunir et de prolonger considérablement la vie humaine en santé.
La détérioration des fonctions cellulaires et organiques associée à l’âge s’accompagne d’une dysrégulation des voies de détection des nutriments et d’une autophagie désactivée. La réactivation du flux autophagique peut prévenir ou améliorer les dysfonctionnements métaboliques liés à l’âge. Les composés non toxiques dotés de la capacité de réduire les niveaux globaux d’acétylation protéique et d’induire l’autophagie ont été classés dans la catégorie des mimétiques de restriction calorique (MRC). Nous montrons ici que l’aspirine ou son métabolite actif salicylate induit l’autophagie en raison de leur capacité à inhiber l’activité acétyltransférase de l’EP300.
Nous démontrons que l’aspirine ne parvient pas à moduler le flux autophagique dans les cellules dépourvues d’EP300 ou dans lesquelles l’EP300 a été conçu pour éviter la liaison de l’aspirine à l’enzyme. Comme confirmation de la nature évolutive conservée de ce processus, nous démontrons que l’aspirine n’ a pas réussi à induire davantage l’autophagie chez les souches de Caenorhabditis elegans déficientes pour l’homologue CBP-1 de l’EP300 ou pour les produits essentiels des gènes de l’autophagie ATG-7 et BEC-1.
D’après les résultats décrits dans le présent document, l’aspirine peut être classée dans la catégorie des MRC. En effet, l’aspirine remplit tous les critères d’un MRC car elle (1) réduit l’acétylation des protéines en raison de sa capacité à inhiber l’activité acétyltransférase de l’EP300, (2) stimule le flux autophagique et (3) n’ a pas d’activité cytotoxique. À ce stade, il reste à déterminer dans quelle mesure l’inhibition de l’EP300 et l’activation de l’autophagie peuvent contribuer efficacement à ces effets d’aspirine qui transcendent apparemment ses effets anti-inflammatoires bien établis.
Les données précliniques suggèrent qu’un dérivé d’aspirine perméable au cerveau peut réduire la neurodégénérescence à médiation tau dans le cas de l’EP300. Cependant, le rôle de l’autophagie n’ a pas été exploré dans ce contexte. Les données épidémiologiques et expérimentales indiquent qu’une absorption nutritionnelle élevée de l’inhibiteur EP300 de la spermidine contrecarre le vieillissement cardiaque, tant chez les humains que chez les rongeurs. De plus, la spermidine réduit l’artériosclérose et la carcinogenèse du côlon chez les modèles murins. Ces effets de la spermidine montrent donc un chevauchement notable avec ceux de l’aspirine, en accord avec l’observation selon laquelle les deux composés inhibent EP300.













 Vers des traitements durables qui fonctionnent à l’intérieur du corps
Vers des traitements durables qui fonctionnent à l’intérieur du corps Les DAMP peuvent établir un lien entre la dysfonction mitochondriale liée à l’âge et l’inflammation chronique
Les DAMP peuvent établir un lien entre la dysfonction mitochondriale liée à l’âge et l’inflammation chronique