Une approche systémique du vieillissement et de la détermination de la durée de vie
En 2014, des scientifiques ont démontré que le sang jeune pouvait rajeunir de vieux tissus en combinant ces deux éléments chez les souris [130]. De plus, les manipulations qui prolongent la durée de vie ciblent un tissu qui pourrait avoir des conséquences bénéfiques sur d’autres [131] [132] [133]. Ces résultats intéressants ont entraîné une recherche active sur les facteurs systémiques circulants, qui pourraient contrôler et coordonner le taux de vieillissement des différents tissus et organes.
L’inflammation chronique liée au vieillissement
« L’inflammaging » est un phénotype pro-inflammatoire observé chez les mammifères vieillissants [134]. Les cytokines jouent un rôle majeur dans la réaction inflammatoire, elles permettent la communication entre les cellules immunitaires et les cellules somatiques. Leur réponse chronique pourrait être partiellement responsable de la vulnérabilité, le développement des maladies liées au vieillissement, et le déclin du vieil âge [135] [136].
De multiples causes sont examinées : l’accumulation de lésions dans les tissus pro-inflammatoires au fil du temps, la sécrétion de cytokines pro-inflammatoires par des cellules sénescentes, le système immunitaire défaillant qui ne parvient pas à éliminer les éléments pathogènes et les cellules-hôtes défaillantes, la mauvaise gestion de l’apoptose en raison du dysfonctionnement des mitochondries, une mauvaise réponse de l’autophagie et une accumulation des protéines endommagées qui entraîne une réaction inflammatoire, un changement des niveaux d’hormones qui régulent la production de cytokine (dont la testostérone) et l’activation améliorée du facteur de transcription NF-κB.
Ces altérations donnent lieu à une amélioration de la production d’interleukine 1β (IL-1β), un facteur de nécrose tumorale [101] [134] qui entraîne une libération du TNF-α. L’interleukine-6 (IL-6) est libérée par le TNF-α et l’interleukine IL-1β.
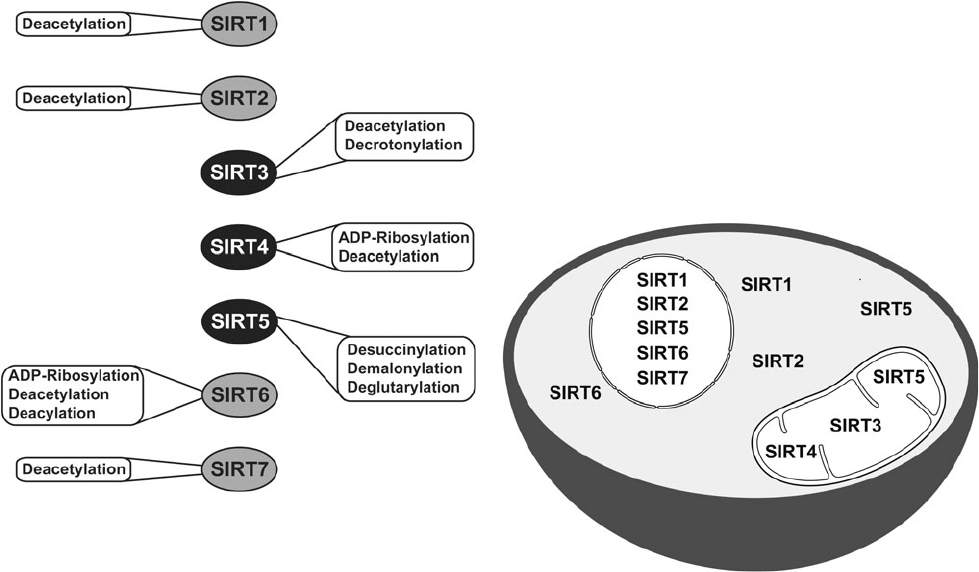
Le facteur nucléaire –κB (NF-κB) participe à la réponse immunitaire et à la réponse au stress. Il contrôle de nombreux gènes liés à l’inflammation et est considéré comme un responsable moléculaire de l’inflammaging. L’inhibition de NF-κB prévient des caractéristiques du vieillissement avancé chez différentes souris modèles [137] [138]. De plus, l’inhibition de la protéine NF-κB entraîne un rajeunissement phénotypique des tissus [139]. Il faut noter que l’expression de la sirtuine SIRT-1 inhibe le NF-κB.
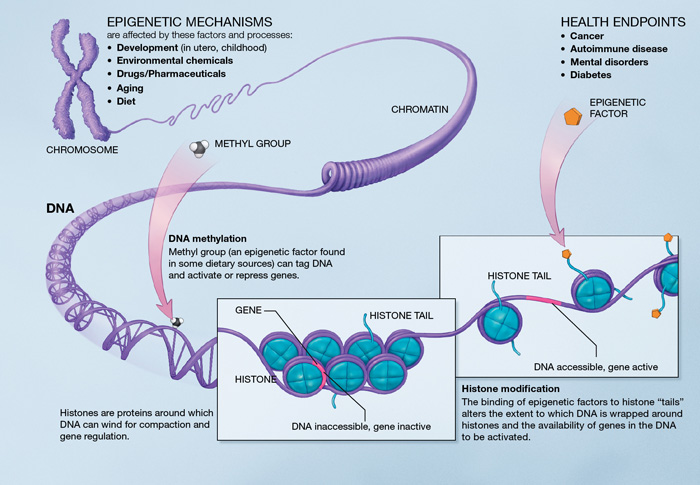
Les sirtuines pourraient avoir également une influence sur l’inflammaging. Des études ont révélé qu’en procédant à la désacétylation des histones et des composants des voies de signalisation comme le NF-κB, la sirtuine SIRT1 régule négativement les gènes associés à l’inflammation. La diminution des niveaux de SIRT-1 est associée au développement et à la progression de nombreuses maladies inflammatoires, et l’activation pharmacologique du SIRT1 éviterait des réactions inflammatoires chez les souris [141] [142] [143]. Les sirtuines SIRT-2 et 6 réguleraient négativement la réponse inflammatoire au moyen de la désacétylation des sous-unités du NF-κB et de la répression transcriptionnelle de leurs gènes cibles [60] [144].
Des études sur le facteur de dégradation d’ARNm AUF-1 apportent une preuve in vivo du lien entre le vieillissement et l’inflammation. L’AUF-1 contrôle la dégradation l’ARNm de cytokine et intervient dans la régulation de la limite de la réaction inflammatoire. Les souris qui ont une carence en AUF-1 présentent une sénescence cellulaire prononcée et un vieillissement prématuré des phénotypes. Il faut noter que l’AUF-1 contribue également au maintien de la longueur des télomères [145].
Une fois de plus, l’inflammation liée au vieillissement peut être interprétée comme un mécanisme de défense qui finit par devenir nocif avec le temps. De faibles niveaux de réponse inflammatoire seraient favorables à la réparation et à la régénération tissulaire par l’activation du système immunitaire ; cependant, des niveaux plus élevés pourraient aggraver les lésions. L’inflammation liée au vieillissement pourrait également fragiliser la fonction des cellules souches [146].

ARNmis
Les micro-ARN (ARNmis) sont un groupe non codant de petits ARNs. Ils peuvent moduler l’expression génétique en ciblant l’ARNm avec une séquence complémentaire. Il faut noter que les ARNmis peuvent contrôler la durée de vie chez les invertébrés [147] [148] [149] en interagissant avec les composants des réseaux de la longévité ou en régulant les capacités régénératrices des cellules souches. Ils pourraient devenir un biomarqueur intéressant du vieillissement et des maladies qui y sont liées, car la caractérisation des ARNmis révèle des altérations considérables dans leurs niveaux d’expression [143].
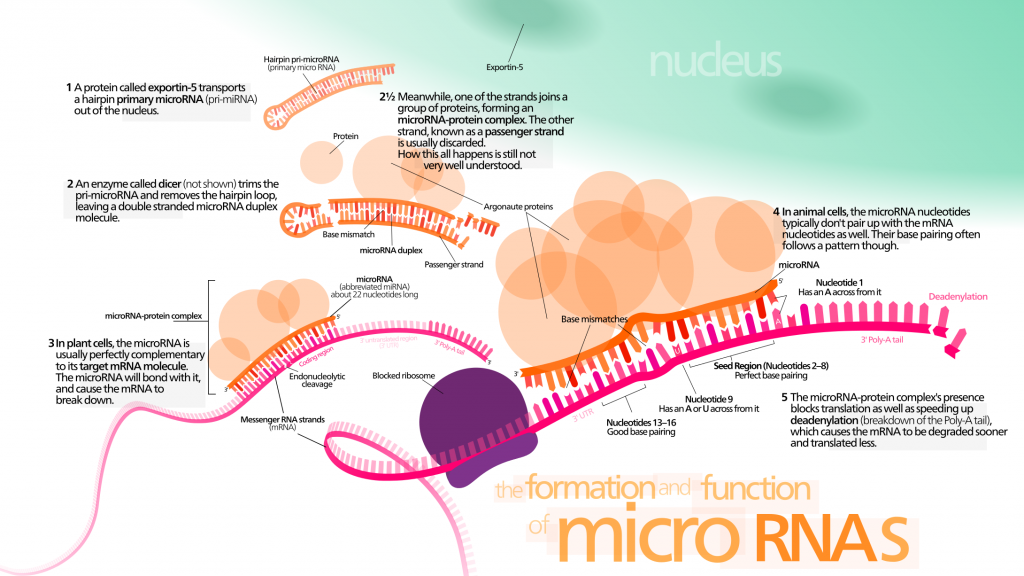
Des ARNmis sans cellules ont attiré l’attention des biologistes, car ils pourraient apporter des informations utiles pour un diagnostic précoce du cancer et d’autres maladies liées au vieillissement. Les ARNmis circulants seraient des facteurs systémiques majeurs du vieillissement. Ils sont connus pour participer à de nombreux aspects du processus de vieillissement :
- La longueur des télomères Les ARNmis participerait à la régulation de la longueur des télomères. De nombreux ANRmis prennent pour cible les ARNm impliqués dans la maintenance des télomères [153]. De plus, chez les humains, l’ANRmi miR-138 est associé négativement au niveau des télomérases reverse transcriptase (hTERT) [154].
- La sénescenceIl existe une preuve qui montre que les ARNmis régulent la sénescence cellulaire et contrôlent l’expression des protéines p53 et p21 [155]. La SIRT-1 pourrait désacétyler et contrôler l’activité de la protéine p53. Le mir -34a est une p53 induite par l’ARNmi, qui peut réguler le niveau d’expression du SIRT-1 chez les souris [156]. Le lien entre la SIRT-1, la p53, et le mir -34 α pourrait représenter un circuit de régulation de la sénescence. En outre, au sein des modèles de sénescence induits par le stress et les oncogènes, la protéine p21 est identifiée comme étant la cible de plusieurs ARNmis [157] [158]. Il faut noter que les niveaux de la famille des miR-106b sont réduits au sein des cellules sénescentes. Le gène mir -106a prend notamment pour cible la protéine p21 ARNm [159] et empêche la sénescence cellulaire, indépendamment du fait que la protéine p53 soit inhibée ou non. D’autres membres de la famille des miR-106b favorisent le cycle cellulaire en prenant pour cible la protéine p21 [158] [160]. Les ARNmis sont suspectés de se trouver au point de croisement entre la sénescence et les maladies liées au vieillissement.
- L’épuisement des cellules souches Certaines ARNmis régulent les propriétés des cellules souches et pourraient ainsi et entretenir la capacité de renouvellement des cellules.
L’inflammation Les ARNmis participerait à la régulation de l’inflammation par l’intermédiaire d’interactions avec la protéine NF-κB [161] [162]. Un petit nombre relatif d’ARNmis semble participer à la régulation de l’inflammation: leurs prototypes sont les micro-ARN155, micro-ARN7 et le micro-ARN 146a [163] [164]. Dans des conditions physiologiques, leur transcription est à un niveau de base, mais les résultats de signalisation pro-inflammatoires ont pour conséquence une forte co-induction de leur expression au moyen d’un mécanisme dépendant de NF-kB.
L’atrophie thymique et son rôle dans l’immunosénescence
Le thymus est un organe qui soutient le développement et la différenciation des lymphocytes T. Les lymphocytes T jouent un rôle décisif dans le système immunitaire adaptatif, qui permet au corps de répondre spécifiquement aux corps étrangers.
Dans les années 1980, Steinman a démontré que la fonction thymique diminue progressivement dès la première année de la vie [166] et que le thymus s’atrophie au cours du vieillissement. Cette involution a pour conséquence une efficacité diminuée du développement des lymphocytes T, et une émigration amoindrie des lymphocytes T naïfs [168]. De ce fait, le système immunitaire adaptatif se dégrade tandis que le système immunitaire inné se renforce.
Alors que le système immunitaire devient moins efficace avec l’âge, l’incidence de maladies telles que les maladies opportunistes, l’auto-immunité ou le cancer augmente [169] [170]. Selon M. R. Dowling et P. D. Hodgkin, ce comportement visiblement néfaste pourrait avoir été sélectionné pour renforcer la lutte contre les infections et ainsi éviter les réactions contre soi-même. La dégradation associée au vieillissement au sein de la fonction immunitaire pourrait ainsi être un effet secondaire regrettable de ce processus de sélection.
Le rôle de l’hypothalamus
L’hypothalamus communique avec de nombreux tissus au moyen de réseaux hormonaux et neuronaux. Il coordonne les réactions métaboliques et comportementales aux stimuli nutritionnels et environnementaux chez les mammifères. Il pourrait jouer un rôle majeur dans le vieillissement systémique et dans le contrôle de la longévité [172].
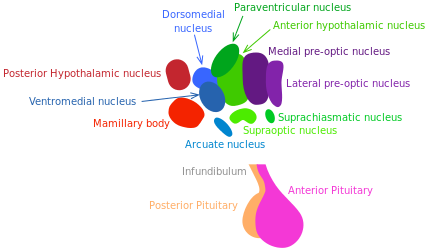
La somatostatine et l’hormone de croissance (GH), un libérateur d’hormones qui sont produites par les neurones hypothalamiques, stimulent et inhibent respectivement la libération de l’hormone de croissance. Il est établi que la voie GH/IGF-1 (régulateur de croissance, réparation tissulaire et métabolique) contrôle la longévité chez les mammifères [173] (voir « Les voies de détection des nutriments »).
Les réactions inflammatoires activent la NF-κB dans l’hypothalamus, déclenchant une voie de signalisation qui réduit la production du GnRH par les neurones. La dégradation de cette hormone pourrait accentuer de nombreux changements systémiques liés au vieillissement comme la fragilité osseuse, la faiblesse musculaire, l’atrophie de la peau, et la neurogénèse amoindrie.
En 2013, Satoh et al. ont démontré que la surexpression transgénique SIRT-1 spécifique au cerveau des souris est liée à un prolongement de la durée de vie. De façon plus intéressante, ces souris présentent des phénotypes cohérents avec un retard du vieillissement [175]. Ces phénotypes révèlent une activité neuronale améliorée, notamment dans le noyau hypothalamique dorso-médian et latéral. Dans cette expérience, des informations complémentaires confirment le rôle du signalement du médiateur SIRT-1 dans ces zones de l’hypothalamus quant au prolongement de l’espérance de vie. Les résultats de Satoh et al. suggèrent que l’activité améliorée de la sirtuine SIRT-1 au sein du noyau hypothalamique latéral et dorso-médian joue un rôle essentiel dans la régulation de la longévité chez les mammifères.
















 Au cours des dernières décennies des scientifiques ont prolongé la vie humaine en soignant des maladies, et en améliorant la santé des citoyens… Aujourd’hui, dans notre société moderne, l’espérance de vie est comprise entre 65 et 85 ans.
Au cours des dernières décennies des scientifiques ont prolongé la vie humaine en soignant des maladies, et en améliorant la santé des citoyens… Aujourd’hui, dans notre société moderne, l’espérance de vie est comprise entre 65 et 85 ans. Le prolongement de la durée de vie est aujourd’hui dû à l’amélioration de la santé publique, au combat contre le cancer et les maladies infantiles (qui augmentent avec l’âge moyen de décès) et non pas, selon l’étude, en raison du phénomène de vieillissement. Les scientifiques expliquent cette horloge biologique en comparant différentes espèces: une souris vit 1000 jours, un chien 5000 jours et un humain 29 000 jours. Ces différences sont biologiques, et en comprendre la cause est une façon de prolonger notre durée de vie.
Le prolongement de la durée de vie est aujourd’hui dû à l’amélioration de la santé publique, au combat contre le cancer et les maladies infantiles (qui augmentent avec l’âge moyen de décès) et non pas, selon l’étude, en raison du phénomène de vieillissement. Les scientifiques expliquent cette horloge biologique en comparant différentes espèces: une souris vit 1000 jours, un chien 5000 jours et un humain 29 000 jours. Ces différences sont biologiques, et en comprendre la cause est une façon de prolonger notre durée de vie.



featured image")
 Pourquoi est-il important d’être rapide et précis?
Pourquoi est-il important d’être rapide et précis?

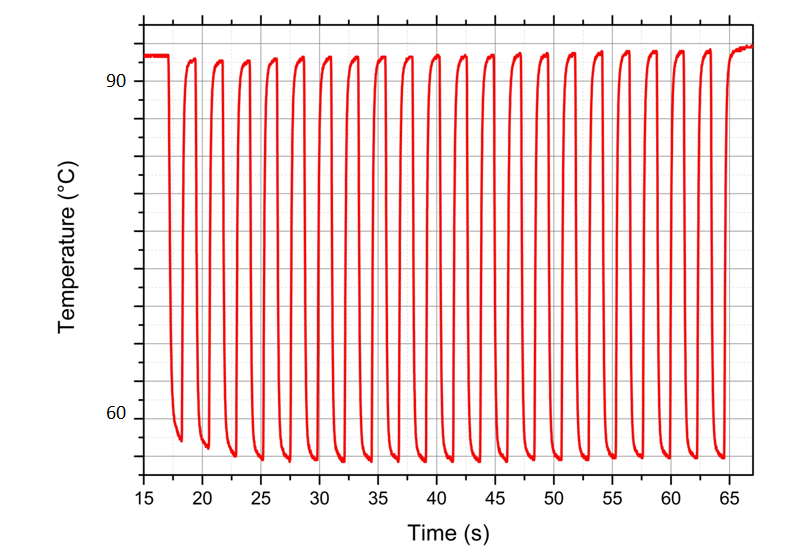
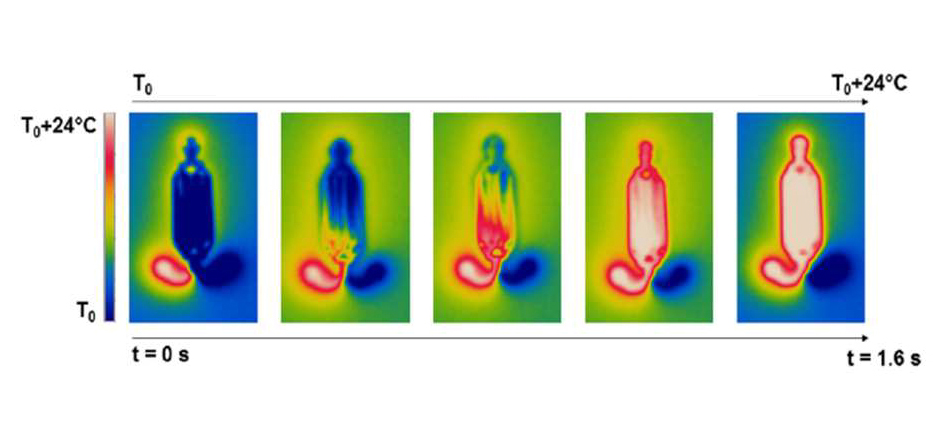
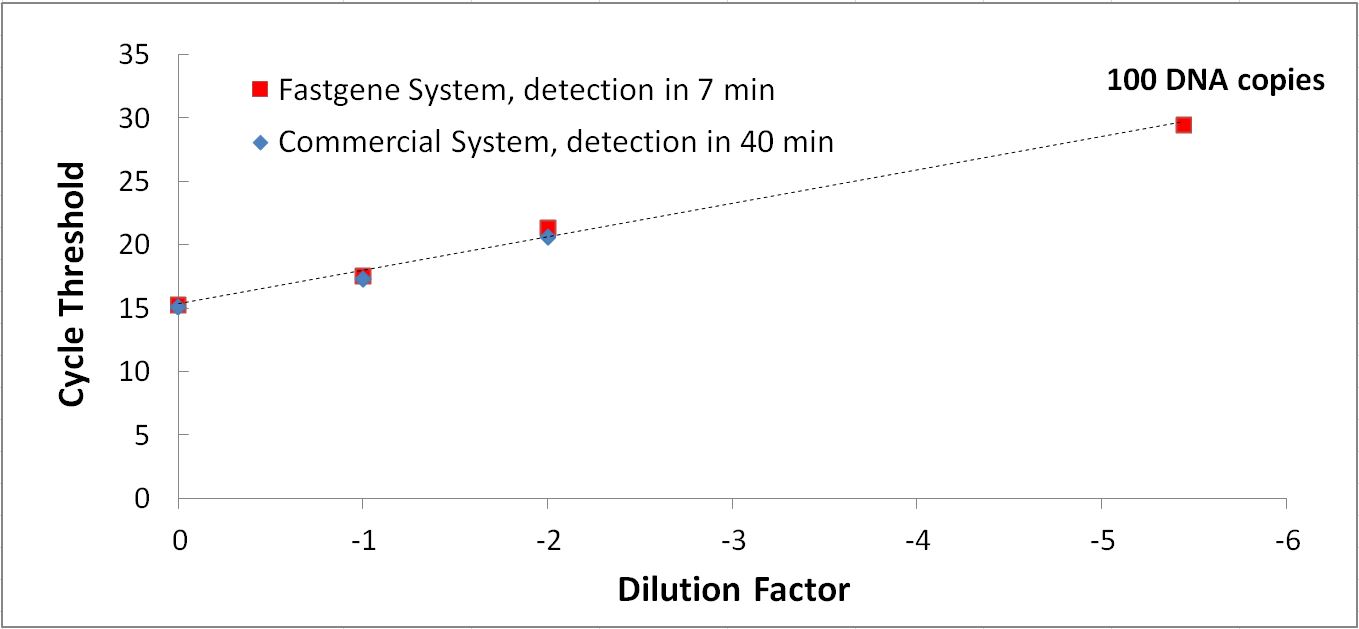
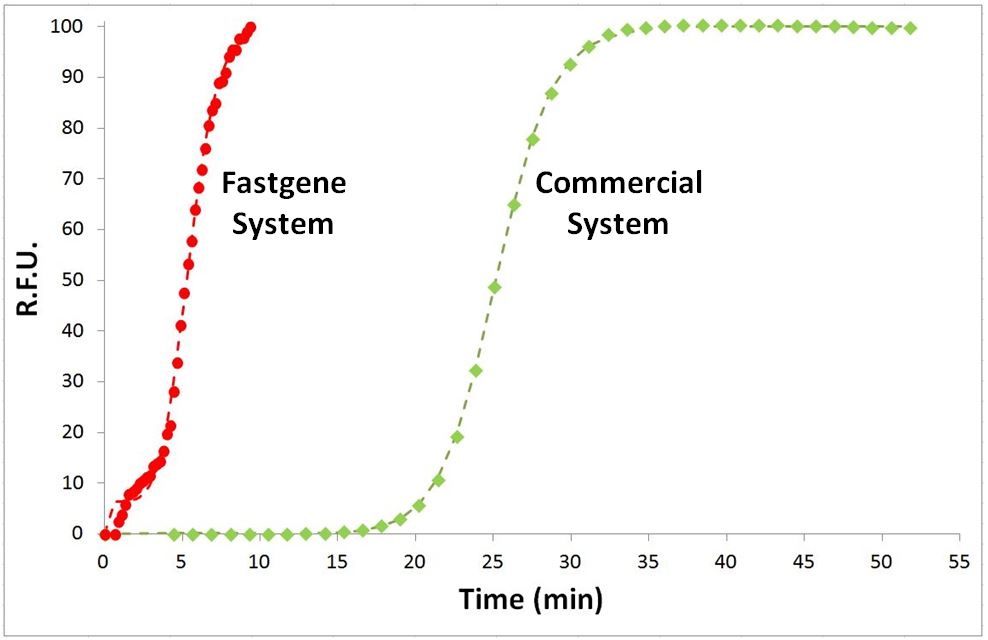 La détection moléculaire de FastGene comparée à un système de pointe commercialisé : le système FastGene permet une détection six fois plus rapide.
La détection moléculaire de FastGene comparée à un système de pointe commercialisé : le système FastGene permet une détection six fois plus rapide.