


Qu’est-ce que le stress cellulaire ?
Dans un organe, il existe une balance entre la croissance et la mort des cellules qui le composent, qu’on appelle homéostasie tissulaire [1]. Chaque cellule est capable de grandir pour s’adapter à une condition physiologique ou pathologique, de changer de conformation ou potentiellement de se multiplier. Elle sera tout aussi capable d’enclencher un mécanisme de mort programmée si le besoin s’en fait sentir ou que la pression à laquelle elle est soumise est trop importante.
Lors d’un stress cellulaire, cette homéostasie est en danger et la cellule aura plusieurs possibilités pour y répondre en fonction du type de stress. Globalement, tant que le stimulus est gérable, la cellule va s’adapter. Dans le cas contraire, elle devra mettre en place des mécanismes de réponse qui peuvent varier [2, 3].
La réponse Heat Shock
Comme son nom l’indique, cette voie a été découverte par l’étude des cellules soumises à un stress cellulaire d’origine thermique [4]. Il est maintenant admis que d’autres stress peuvent l’activer mais le nom est resté. Cette réponse consiste à augmenter le nombre et/ou l’activité des protéines chaperons afin d’éviter que les protéines synthétisées par la cellule ne se replient mal. Le repliement des protéines est indispensable à leur bonne fonction, et il n’est pas rare que certaines maladies résultent d’une protéine synthétisée correctement, mais dont la conformation finale ne permet pas un fonctionnement adéquat. Dans le cas d’un stress cellulaire, les protéines peuvent perdre leur forme finale, se déplier, se replier ou même s’agréger entre elles, alors qu’elles ne devraient pas. La réponse heat shock lutte contre cet effet, afin de permettre à la cellule de continuer à fonctionner dans des conditions non optimales. Cette réponse confère à la cellule une protection connue sous le nom de thermotolérance [5, 6, 7].

Dans un premier temps, la transcription et traduction des gènes est mise en pause pour éviter la synthèse d’un trop grand nombre de protéines mal repliées que la cellule ne pourrait pas gérer. Dans un second temps, des facteurs de transcription vont entrer en jeu et activer la synthèse de molécules spécifiques, les heat shock factors (HSF) [8]. Nous avons à notre disposition 4 isoformes de ces facteurs, HSF1, 2, 3 et 4 (oui, les scientifiques ne sont pas toujours très inventifs !), chacune ayant un rôle différent, mais dont la plus étudiée pour sa fonction dans la réponse au stress cellulaire est HSF1 [9]. Dans des conditions normales, HSF1 garde sa forme inactive, un monomère cytoplasmique, en interaction avec des protéines chaperons [10].
Quand la cellule est soumise à un stress, on observe une accumulation de protéines mal repliées qui vont entrer en compétition avec HSF1 pour se lier aux protéines chaperons. A partir de ce moment là, HSF1 est libéré, va s’associer avec ses homologues pour former un trimère actif qui va aller se lier à l’ADN et promouvoir l’expression spécifique de certains gènes, notamment ceux codant pour les heat shock proteins (HSP).
Les HSP peuvent être exprimées constitutivement, comme Hsp90 qui est l’une des protéines chaperons maintenant HSF1 sous sa forme inactive. D’autres vont être spécifiquement exprimées lors d’un stress cellulaire, comme Hsp27 ou Hsp70 [5]. Elles auront différentes actions mais vont généralement aller dans le sens de la survie cellulaire. Elles peuvent se fixer aux protéines mal repliées [11] pour éviter leur agrégation, garantir leur fonction et maintenir l’intégrité du cytosquelette [12]. Elle permettent également le blocage de la mort cellulaire programmée en inhibant certaines protéines pro-apoptotiques comme les caspases ou le cytochrome C [13, 14, 15].
La réponse Unfolded Protein (UPR)
Comme nous l’avons vu précédemment, le repliement correct des protéines est indispensable à leur fonction, mais pour qu’une protéine travaille convenablement, elle doit également subir toutes sortes de modifications post-traductionnelles comme la glycosylation, la formation de ponts disulfure, l’oligomérisation… Tout cela a lieu dans un organite spécifique, le réticulum endoplasmique. Certains chocs, comme la restriction calorique ou la diminution de l’apport en oxygène, peuvent engendrer un stress du réticulum endoplasmique, résultant en un afflux de protéines mal repliées, mal modifiées, un mécanisme connu sous le nom de « unfolded protein response » (UPR) [16, 17]. Cette voie de signalisation est activée via des protéines spécifiques qui sont, pour la plupart, présentes sous forme inactive dans des conditions physiologiques, et vont être activées lors du stress cellulaire. On compte parmi elles IRE-1, PERK et ATF6, qui auront pour rôle de se fixer sur l’ADN et d’encourager la synthèse de certains gènes codant notamment pour des protéines chaperons et des anti-oxydants [16].
L’UPR est soumis à une balance délicate liée à la capacité du réticulum à replier correctement les protéines et à gérer celles qui ne le sont pas. Lorsque l’UPR est apte à améliorer cette capacité du réticulum pour faire face au stress, via la synthèse de facteurs et de protéines comme IRE-1, elle encouragera la survie cellulaire. Si le réticulum est dépassé, l’UPR favorisera plutôt la mort cellulaire par des mécanismes encore peu connus.
La réponse génotoxique
L’ADN contenu dans le noyau de nos cellules est généralement sous forme double brin, comme une hélice. Lors de certains événements, notamment la transcription et la réplication de l’ADN, il se retrouve sous forme mono brin : les enzymes qui vont permettre ces phénomènes ont besoin d’accéder à l’information contenue dans l’ADN et vont séparer les deux brins afin de les copier ou les transcrire. C’est à ce moment là que l’ADN est le plus sensible aux stress comme l’irradiation, les ultraviolets ou les traitements anti-cancer (parfois qualifiés d’intercalants de l’ADN). Il va être possible de casser l’ADN soit sur un brin (single-strand break ou SSB), soit sur les deux brins (double-strand break ou DBS) [18]. Nos cellules sont capables de réparer dans une certaine mesure ces cassures mais parfois, elles induisent une signalisation différente, en particulier par le biais de p53 (un facteur de transcription qui régule l’apoptose, l’autophagie et le cycle cellulaire) et NF-κB (une cytokine pro-inflammatoire et pro-apoptotique).

Là encore, tout est affaire de balance. Lorsque les dommages de l’ADN sont réparables, les voies de signalisation enclenchées seront en faveur de la survie cellulaire. Si les cassures sont trop importantes, p53 et NF-κB entrent en jeu et vont activer la voie de mort de la cellule.
Ces voies de signalisation sont extrêmement complexes, interconnectées et encore étudiées à ce jour.
La réponse au stress oxydant
Les espèces réactives de l’oxygène (aussi appelée ROS « reactive oxygen species ») sont l’une des cause principales de stress cellulaire. La survie d’une cellule dépend d’un équilibre entre oxydants (oxygène et ROS) et anti-oxydants (enzymes et protéines telles que le glutathion, la catalase ou les superoxyde dismutases) qui peut être dysfonctionnel [19]. La plupart du temps, la cellule est en capacité de lutter contre le stress oxydant et de favoriser sa survie mais, comme pour les précédents stress cellulaires évoqués, il arrive que la balance oxydants:anti-oxydants soit trop dérégulée pour que la cellule la corrige. Elle entrera alors dans des processus d’apoptose ou de nécrose [19, 20, 21].
Les plus connues des ROS sont le peroxyde d’hydogène (H202, aussi appelé eau oxygénée), les radicaux hydroxyl (OH•) et l’oxyde nitrique (NO•). Ils ont des origines variées, intra- ou extracellulaires. La plus grande source d’oxydants intracellulaires est notre mitochondrie et sa chaine respiratoire : lorsqu’elle produit de l’énergie, elle produit également des ROS qui sont généralement traitées grâce aux superoxyde dismutases. En plus de cette source intracellulaire, notre mode de vie influence grandement la génération de ROS. On retrouve parmi les causes externes, l’exposition prolongée aux UV, la pollution, les pesticides, le tabac, l’alcool, une alimentation déséquilibrée, trop de sport, le stress, ou encore une déficience nutritionnelle en un ou plusieurs antioxydants.
La réponse au stress oxydant allie une réponse spécifique et des mécanismes communs aux autres stress, comme l’activation de la synthèse de protéines chaperons [22] ou de NF-κB et p53 [23]. Il n’est pas encore très clair comment l’induction de l’apoptose a lieu mais il semblerait que l’H2O2 active un système appelé Fas-FasL, qui induirait à son tour l’activation des caspases [24]. NO•, quant à lui, semble être capable d’inactiver certaines enzymes anti-oxydantes telles que les superoxyde dismutase [25]. De manière intéressante, les ROS pourraient également activer un mécanisme de mort cellulaire différent, la nécrose. Cela se produit grâce à l’inactivation des caspases, des molécules normalement pro-apoptotiques et dont l’absence de stimulation va obliger la cellule à se tourner vers une autre forme de mort. L’engagement vers la nécrose peut également s’expliquer par une diminution d’ATP dans la cellule, inhérente à un dommage au niveau des mitochondries par un trop grand nombre d’oxydants [26]. Enfin, un dernier mode peut être enclenché, celui de l’autophagie, bien que les mécanismes y menant ne soit pas encore clairs [27].
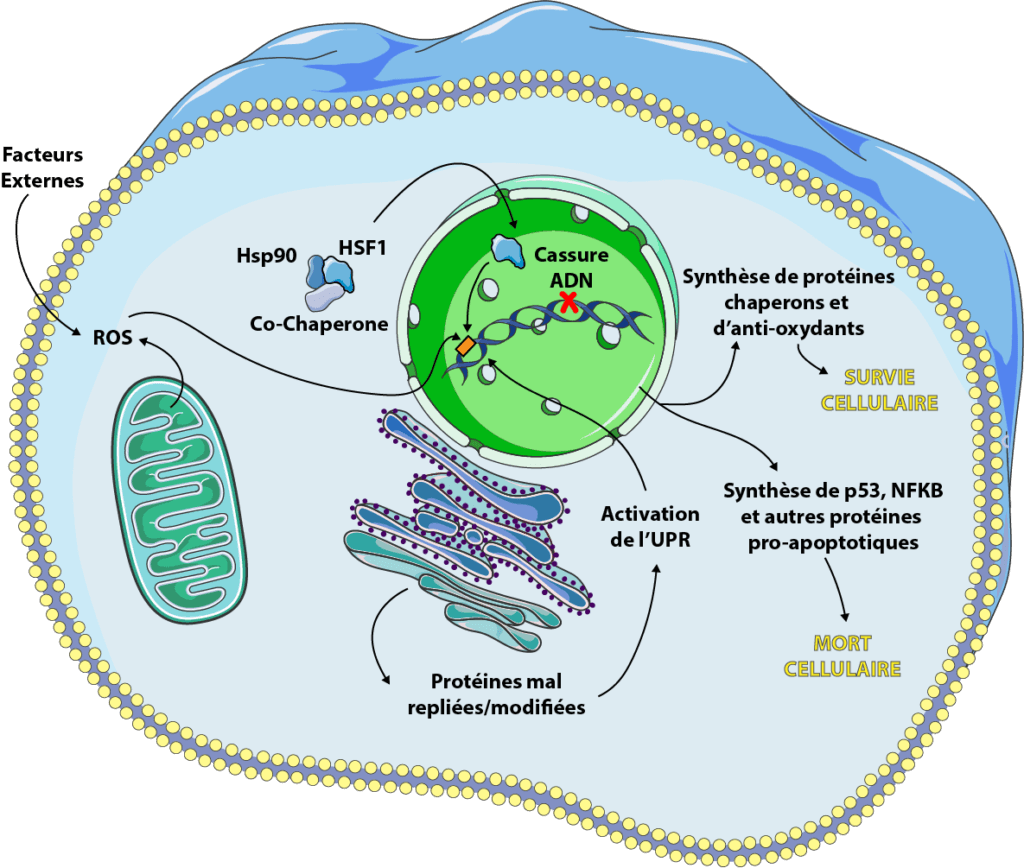
Stress cellulaire et vieillissement
Parmi les stress que nous avons détaillés, nombreux sont ceux qui s’aggravent avec l’âge. En effet, beaucoup de maladies, notamment le diabète, les maladies neurodégénératives (Parkinson et Alzheimer en première ligne) ou les cancers, sont en partie causées par une dérégulation de la protéostasie et de l’équilibre oxydants:antioxydants (voir dossier causes du vieillissement). Il est, par exemple, prouvé que la synthèse de HSP augmente lorsque l’on vieillit [28] et qu’elle est souvent couplé à une dérégulation de l’expression de certains gènes via le système de signalisation de HSF1 qui fonctionne de moins en moins bien [29]. Globalement, le stress, en particulier oxydant, est intimement lié à l’inflammation (via la production de cytokines, comme NF-κB) et lorsqu’elle devient chronique, elle est associée à une augmentation des ROS. C’est une sorte de boucle qui s’auto-entretient et accélère notre vieillissement. Le stress cellulaire est donc à la fois une cause et une conséquence des pathologies cellulaires et du vieillissement.
Quelques définitions
Cytokines : ce sont des molécules synthétisées par nos cellules immunitaires qui permettent la communication entre les cellules. Elles sont souvent associées à l’inflammation car ce sont les actrices majeures de ce processus.
Protéines chaperons : ce sont de petites protéines dont le rôle est d’accompagner les protéines en cours de synthèse jusqu’à ce qu’elles acquièrent leur conformation finale. Elles empêchent également l’agrégation protéique.
Facteur de transcription : c’est une molécule qui va pouvoir se fixer sur des zones spécifiques de l’ADN, appelées promoteurs, qui régulent l’expression des gènes. Ils peuvent avoir un rôle activateur ou inhibiteur de la transcription.
Monomère/dimère/trimère protéique : lorsque les protéines se forment, pour arriver à leur fonction finale, elles ont parfois besoin de se lier entre elles. Lorsqu’elles sortent du réticulum, elles sont sous forme de monomère (c’est à dire une seule chaine peptidique qui forme une seule protéine). Dans le cytoplasme, elles peuvent ensuite se lier avec une, deux ou plusieurs protéines formant des dimères (deux chaines), des trimères (trois chaines)… On distingue les homodimères, qui sont l’association de deux protéines identiques, des hétérodimères, qui résultent de la liaison de deux isoformes d’une même protéine ou de deux protéines différentes.
Apoptose : c’est le mécanisme enclenchée par la cellule elle-même lorsqu’elle décide de mourir. On distingue l’apoptose de la nécrose par les voies de signalisation impliquées dans la mise en place du processus de mort cellulaire.
Autophagie : c’est le mécanisme par lequel une cellule est capable de se débarrasser de certains organites dysfonctionnels, en les digérant ou en les expulsant de son cytoplasme. Lorsque la cellule arrive à gérer ces dysfonctionnements, cette voie est pro-survie. En revanche, lorsqu’il y a trop d’organites qui ne marchent plus ou que l’autophagie n’est pas suffisante à les traiter, cette voie sera pro-mort.
Tout notre dossier Stress cellulaire et vieillissement :
Stress cellulaire et vieillissement, binôme obligatoire ?

Bien que communément acquis par la communauté scientifique comme un facteur de risque et un déterminant de certaines maladies, beaucoup de questionnements subsistent vis à vis du stress cellulaire. Son rôle dans les pathologies liées au vieillissement et le vieillissement lui-même est toujours en cours d’étude.
Partie 1 : Les différents types de stress cellulaires
 Quand on parle de stress cellulaire, nous pouvons faire référence à beaucoup de mécanismes différents, des stress mécanique, toxique, chimique, thermique, osmotique, ionisant… tous participant au vieillissement de nos cellules.
Quand on parle de stress cellulaire, nous pouvons faire référence à beaucoup de mécanismes différents, des stress mécanique, toxique, chimique, thermique, osmotique, ionisant… tous participant au vieillissement de nos cellules.
Partie 2 : Les organites touchés par le stress cellulaire
 Le stress cellulaire est transmis par différents organites dans nos cellules, notamment la mitochondrie, le réticulum et le noyau, pour ne citer qu’eux. Leurs réponses varient et peuvent donner deux options à la cellule stressée: survivre et s’adapter ou mourir.
Le stress cellulaire est transmis par différents organites dans nos cellules, notamment la mitochondrie, le réticulum et le noyau, pour ne citer qu’eux. Leurs réponses varient et peuvent donner deux options à la cellule stressée: survivre et s’adapter ou mourir.
Partie 3 : Comment maîtriser le stress cellulaire
 Afin de ralentir le vieillissement de nos cellules, il est indispensable de maîtriser le stress cellulaire. Dans cette partie, nous ferons le tour des techniques et/ou médicaments actuellement plébiscités pour traiter le stress cellulaire sous toutes ses formes.
Afin de ralentir le vieillissement de nos cellules, il est indispensable de maîtriser le stress cellulaire. Dans cette partie, nous ferons le tour des techniques et/ou médicaments actuellement plébiscités pour traiter le stress cellulaire sous toutes ses formes.
Références
[1] Simone Fulda, Adrienne M. Gorman, Osamu Hori, and Afshin Samali, “Cellular Stress Responses: Cell Survival and Cell Death,” International Journal of Cell Biology, vol. 2010, Article ID 214074, 23 pages.
[2]C. R. Weston and R. J. Davis, “The JNK signal transduction pathway,” Current Opinion in Cell Biology, vol. 19, no. 2, pp. 142–149, 2007.
[3] N. D. Perkins and T. D. Gilmore, “Good cop, bad cop: the different faces of NF-κB,” Cell Death and Differentiation, vol. 13, no. 5, pp. 759–772, 2006.
[4] S. Lindquist, “The heat-shock response,” Annual Review of Biochemistry, vol. 55, pp. 1151–1191, 1986.
[5] A. Samali and S. Orrenius, “Heat shock proteins: regulators of stress response and apoptosis,” Cell Stress and Chaperones, vol. 3, no. 4, pp. 228–236, 1998.
[6] A. Samali, C. I. Holmberg, L. Sistonen, and S. Orrenius, “Thermotolerance and cell death are distinct cellular responses to stress: dependence on heat shock proteins,” FEBS Letters, vol. 461, no. 3, pp. 306–310, 1999.
[7] G. C. Li and Z. Werb, “Correlation between synthesis of heat shock proteins and development of thermotolerance in Chinese hamster fibroblasts,” Proceedings of the National Academy of Sciences of the United States of America, vol. 79, no. 10, pp. 3218–3222, 1982.
[8] L. Pirkkala, P. Nykänen, and L. Sistonen, “Roles of the heat shock transcription factors in regulation of the heat shock response and beyond,” FASEB Journal, vol. 15, no. 7, pp. 1118–1131, 2001
[9] A. Shabtay and Z. Arad, “Reciprocal activation of HSF1 and HSF3 in brain and blood tissues: is redundancy developmentally related?” American Journal of Physiology, vol. 291, no. 3, pp. R566–R572, 2006.
[10] I. Shamovsky and E. Nudler, “New insights into the mechanism of heat shock response activation,” Cellular and Molecular Life Sciences, vol. 65, no. 6, pp. 855–861, 2008.
[11] F. U. Hartl and M. Hayer-Hartl, “Molecular chaperones in the cytosol: from nascent chain to folded protein,” Science, vol. 295, no. 5561, pp. 1852–1858, 2002.
[12] C. G. Concannon, A. M. Gorman, and A. Samali, “On the role of Hsp27 in regulating apoptosis,” Apoptosis, vol. 8, no. 1, pp. 61–70, 2003.
[13] J.-M. Bruey, C. Ducasse, P. Bonniaud et al., “Hsp27 negatively regulates cell death by interacting with cytochrome c,” Nature Cell Biology, vol. 2, no. 9, pp. 645–652, 2000.
[14] D. Chauhan, G. Li, T. Hideshima et al., “Hsp27 inhibits release of mitochondrial protein Smac in multiple myeloma cells and confers dexamethasone resistance,” Blood, vol. 102, no. 9, pp. 3379–3386, 2003.
[15] R. Steel, J. P. Doherty, K. Buzzard, N. Clemons, C. J. Hawkins, and R. L. Anderson, “Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with Apaf-1,” Journal of Biological Chemistry, vol. 279, no. 49, pp. 51490–51499, 2004.
[16] M. Schröder and R. J. Kaufman, “The mammalian unfolded protein response,” Annual Review of Biochemistry, vol. 74, pp. 739–789, 2005.
[17] D. Ron and P. Walter, “Signal integration in the endoplasmic reticulum unfolded protein response,” Nature Reviews Molecular Cell Biology, vol. 8, no. 7, pp. 519–529, 2007.
[18] M. Christmann, M. T. Tomicic, W. P. Roos, and B. Kaina, “Mechanisms of human DNA repair: an update,” Toxicology, vol. 193, no. 1-2, pp. 3–34, 2003.
[19] D. Trachootham, W. Lu, M. A. Ogasawara, N. R.-D. Valle, and P. Huang, “Redox regulation of cell survival,” Antioxidants & Redox Signaling, vol. 10, no. 8, pp. 1343–1374, 2008.
[20] K. Niizuma, H. Endo, and P. H. Chan, “Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival,” Journal of Neurochemistry, vol. 109, supplement 1, pp. 133–138, 2009.
[21] M. Genestra, “Oxyl radicals, redox-sensitive signalling cascades and antioxidants,” Cellular Signalling, vol. 19, no. 9, pp. 1807–1819, 2007.
[22] A. M. Gorman, B. Heavey, E. Creagh, T. G. Cotter, and A. Samali, “Antioxidant-mediated inhibition of the heat shock response leads to apoptosis,” FEBS Letters, vol. 445, no. 1, pp. 98–102, 1999.
[23] M. Meyer, R. Schreck, and P. A. Baeuerle, “H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor,” EMBO Journal, vol. 12, no. 5, pp. 2005–2015, 1993.
[24] T. L. Denning, H. Takaishi, S. E. Crowe, I. Boldogh, A. Jevnikar, and P. B. Ernst, “Oxidative stress induces the expression of Fas and Fas ligand and apoptosis in murine intestinal epithelial cells,” Free Radical Biology & Medicine, vol. 33, no. 12, pp. 1641–1650, 2002.
[25] M. Asahi, J. Fujii, K. Suzuki et al., “Inactivation of glutathione peroxidase by nitric oxide. Implication for cytotoxicity,” Journal of Biological Chemistry, vol. 270, no. 36, pp. 21035–21039, 1995.
[26] M. Leist, B. Single, H. Naumann et al., “Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis,” Experimental Cell Research, vol. 249, no. 2, pp. 396–403, 1999.
[27] R. Scherz-Shouval, E. Shvets, E. Fass, H. Shorer, L. Gil, and Z. Elazar, “Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4,” EMBO Journal, vol. 26, no. 7, pp. 1749–1760, 2007.
[28] Macario AJ, Conway de Macario E, Sick chaperones, cellular stress, and disease, N Engl J Med. 2005 Oct 6; 353(14):1489-501.
[29] Sóti C, Csermely P, Molecular chaperones and the aging process. Biogerontology. 2000; 1(3):225-33.
Dr. Marion Tible
Author/Reviewer
Auteure/Relectrice
Marion Tible has a PhD in cellular biology and physiopathology. Formerly a researcher in thematics varying from cardiology to neurodegenerative diseases, she is now part of Long Long Life team and is involved in scientific writing and anti-aging research.
More about the Long Long Life team
Marion Tible est docteur en biologie cellulaire et physiopathologie. Ancienne chercheuse dans des thématiques oscillant de la cardiologie aux maladies neurodégénératives, elle est aujourd’hui impliquée au sein de Long Long Life pour la rédaction scientifique et la recherche contre le vieillissement.
En savoir plus sur l’équipe de Long Long Life
Dr Guilhem Velvé Casquillas

Author/Reviewer
Auteur/Relecteur
Physics PhD, CEO NBIC Valley, CEO Long Long Life, CEO Elvesys Microfluidic Innovation Center
More about the Long Long Life team
Docteur en physique, CEO NBIC Valley, CEO Long Long Life, CEO Elvesys Microfluidic Innovation Center
En savoir plus sur l’équipe de Long Long Life










